Unit – I
Atomic & molecular structure
Atomic Orbitals is the mathematical function which is responsible for the determination of location and wave like behavior of an electron in the atom. Atomic orbital is the region where the electrons are present in the atoms. Hence the orbitals in an atom is identified from their unique values i.e.; n, l, m.
Properties:
- Electron orbits the nucleus as a Standing waves.
- The electron is at the random position means they never stay at a single position.
Molecular Orbital is the mathematical function which is responsible for the determination of location and wave like behavior of an electron in the molecule. In molecular orbital, the electrons are allowed to interact with more than one atomic nucleus at a time.
Linear Combination of Atomic Orbital (LCAO):
The approximate method used to represent molecular orbital is called as the Linear Combination of Atomic Orbital. It is a quantum superposition of atomic orbital and a technique for calculating molecular orbital in quantum chemistry.
Rules for the Linear Combination of Atomic Orbital are:-
- The combining atoms should have the same symmetry along the molecular axis for proper combination e.g. All the sub-orbitals of 2p have same energy but still, the 2pz orbital of an atom can only combine with a 2pz orbital of another atom but cannot combine with 2px and 2py orbital as they have a different axis of symmetry.
- The two atomic orbital will combine to form molecular orbital. Greater is the extend of overlap of atomic orbital; greater will be the nuclear density.
The combining atomic orbital must be of equal energy or approximately same energy.
H2 molecule:
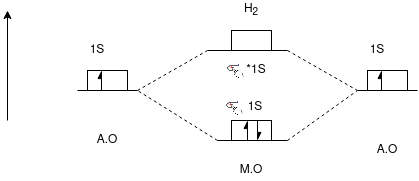
Molecular Orbital Energy Level Diagram of H2 Molecule
(Homonuclear Diatomic Molecule)
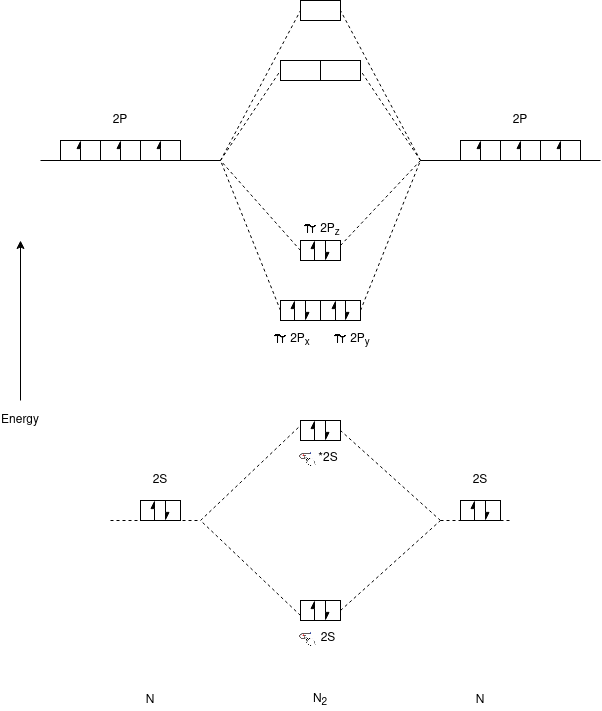
N2 Molecule:-
Electronic Configuration:-
N2 => σ1s2,σ*1s2,σ2s2,σ*2s2,π2px2,π2py2,π*2px2,π*2py2
O2 Molecule:-
Electronic Configuration:-
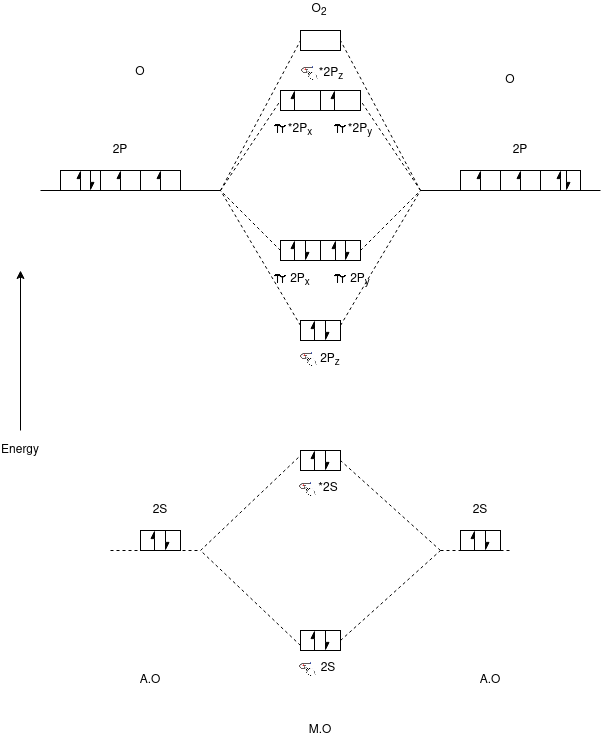 N2 => σ1s2,σ*1s2,σ2s2,σ*2s2,π2px2,π2py2,π*2px1,π*2py1
N2 => σ1s2,σ*1s2,σ2s2,σ*2s2,π2px2,π2py2,π*2px1,π*2py1
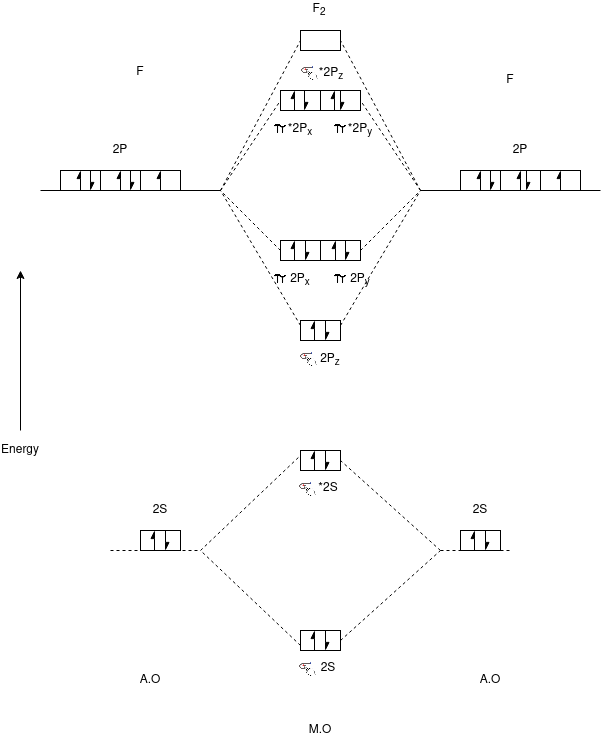
F2 Molecule:-
Electronic Configuration:-
F2=>σ1S2,σ*1S2,σ2S2,σ*2S2,π2P4,π*2P4
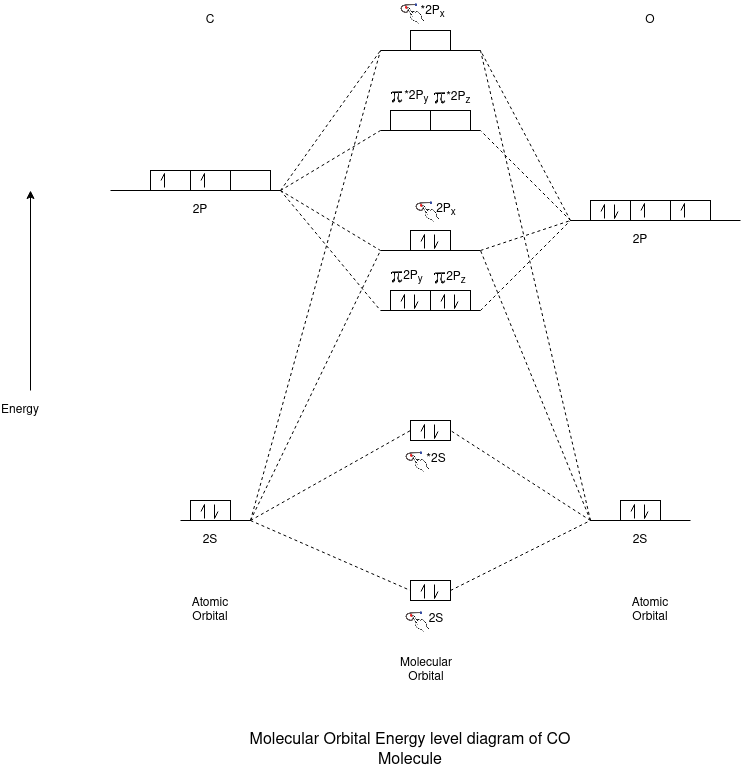
CO Molecule
NO molecule:
NO:-
Electronic Configuration:-
σ1s2,σ*1s2,σ2s2,σ*2s2,π2px2,π2py2,π2pz2,π*2py1,π*2pz0
No of electrons:- 7+8 = 15
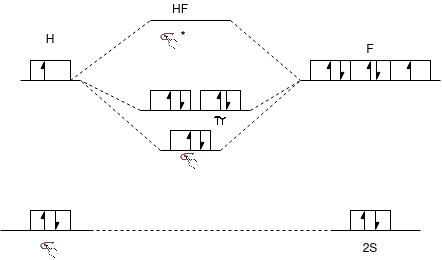
HF Molecule:
Bond order is defined as the value calculated at the difference between number of bonds and anti-bonds. For example, in diatomic nitrogen, N≡N, the bond order is 3; in acetylene, H−C≡C−H, the carbon-carbon bond order is also 3, and the C−H bond order is 1. Bond order and bond length indicate the type and strength of covalent bonds between atoms. Bond order and length are inversely proportional to each other, when bond order is increased, bond length is decreased.
Bond order can be calculated with the following formula:
B.O. = number of bonding electrons – number of anti bonding electrons
2
To determine the bond order between two covalently bonded atoms, follow these steps:
- Draw the Lewis structure.
- Determine the type of bonds between the two atoms.
1- No bond
2- Single bond
3- double bond
4- triple bond
CN-
1) Draw the Lewis structure.
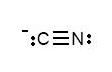
2) Determine the type of bond between the two atoms.
Because there are 3 dashes, the bond is a triple bond. A triple bond corresponds to a bond order of 3.
Polyatomic molecules
If there are more than two atoms in the molecule, follow these steps to determine the bond order:
- Draw the Lewis structure.
- Count the total number of bonds.
- Count the number of bond groups between individual atoms.
- Divide the number of bonds between atoms by the total number of bond groups in the molecule.
NO3-
1) Draw the Lewis structure.
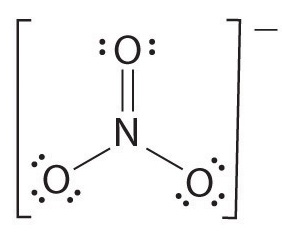
2) Count the total number of bonds.
4
The total number of bonds is 4.
3) Count the number of bond groups between individual atoms.
3
The number of bond groups between individual atoms is 3.
4) Divide the number of bonds between individual atoms by the total number of bonds.
4/3=1.33
The bond order is 1.33
The Butadiene Pi System has zero nodes at its lowest energy molecular orbital.
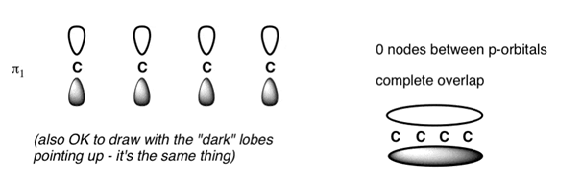
The Butadiene Pi System has three nodes at its lowest energy molecular orbital. The drawn n-p orbital are at alternate phase to each other. This create the a pi system with three nodes.
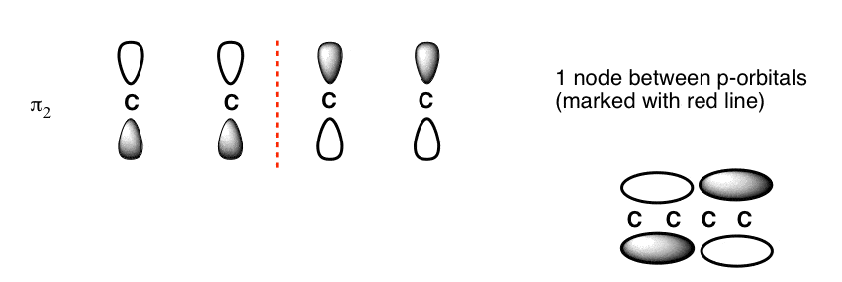
The Butadiene second lowest energy molecular orbital has one node.
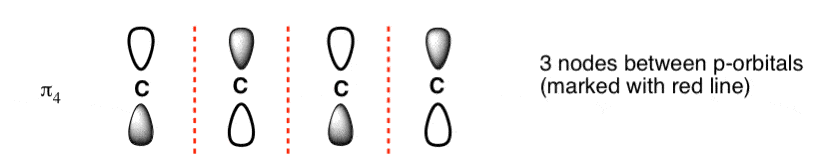
The third lowest energy molecular orbital has two nodes.
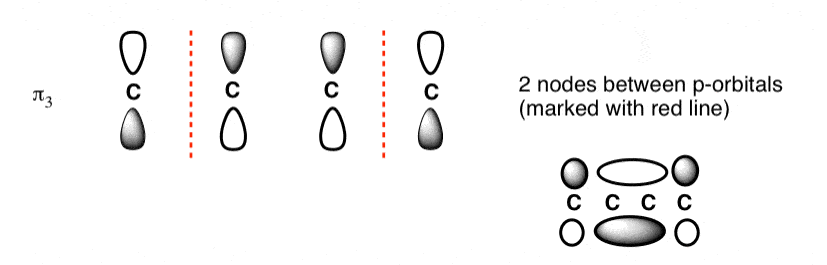
The Full Molecular Orbital Diagram For The Butadienyl System.
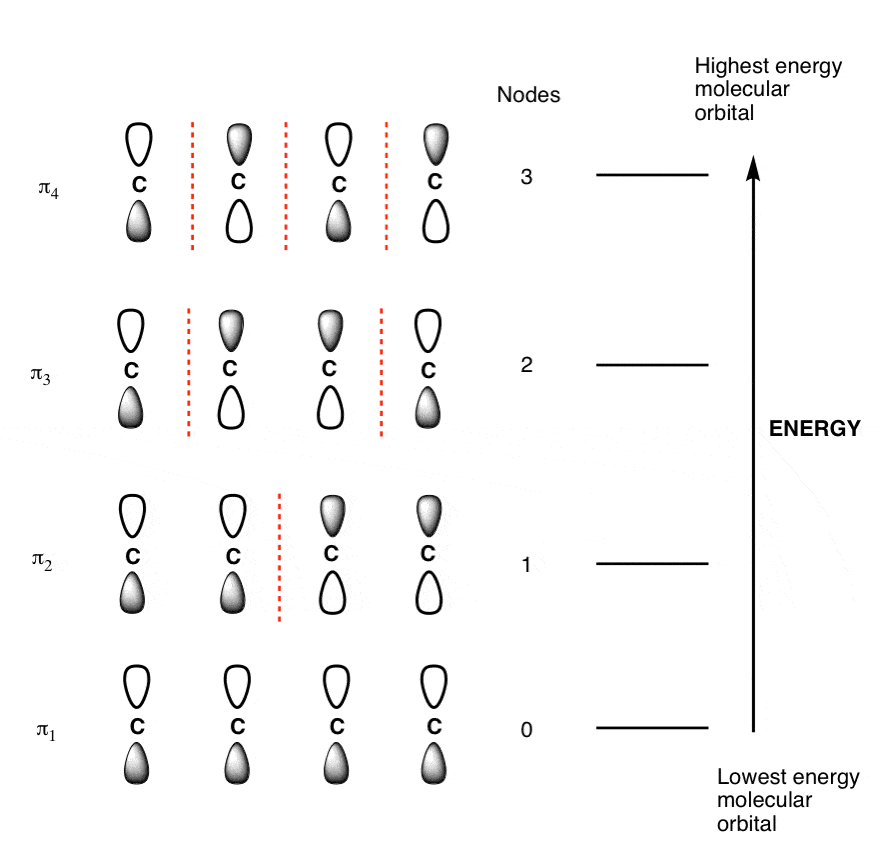
Populating The Molecular Orbitals Of Butadiene With Electrons
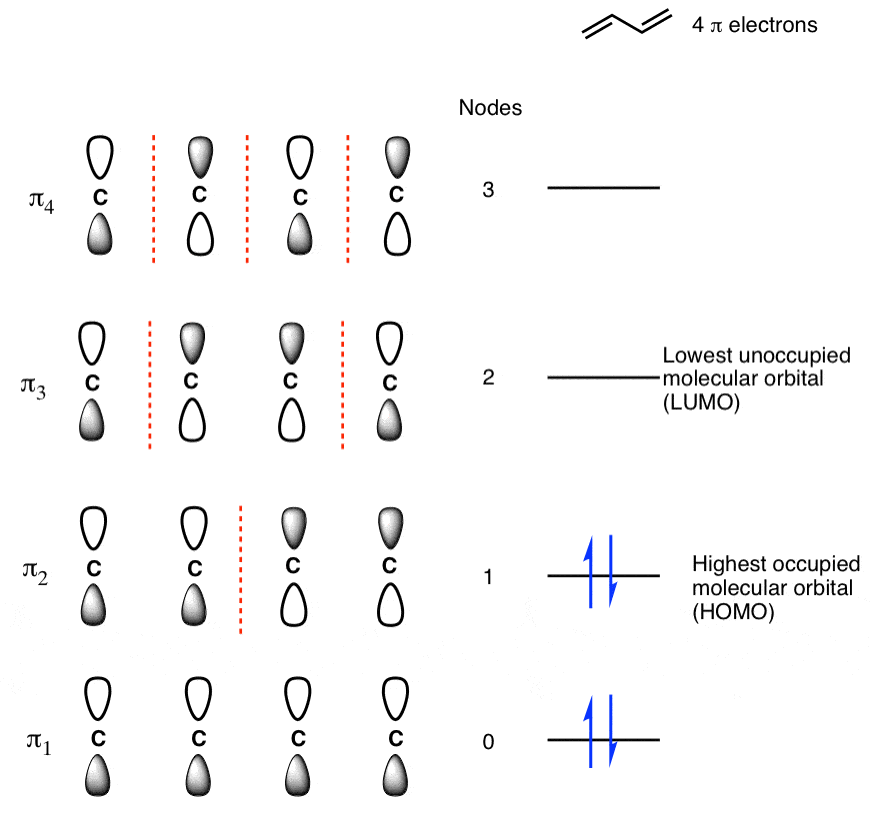
π molecular orbital of benzene:
The Pi molecular orbital diagram for Benzene
In the above increasing energy level the bottom three orbital are all bonding orbital while the top 3 orbital are anti bonding orbital.
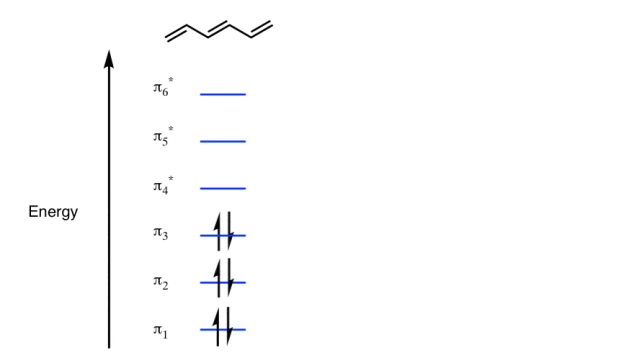
The Benzene System has zero nodes at its lowest energy molecular orbital.
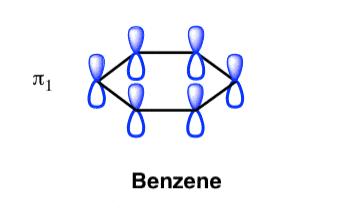
- All p orbital are aligned with phases pointing in the same direction.
- Nodes are absent between orbital.
- In this orbital, electrons are delocalized over the length of molecule, resulting in greatest lowering of energy.
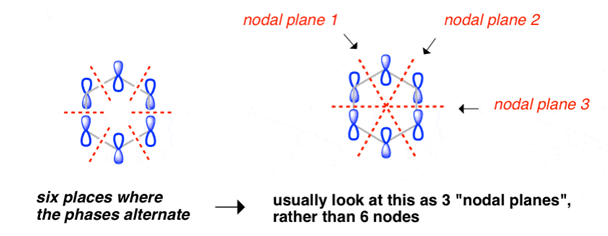
Benzene Has Nodal Planes. The Maximum Energy Level Has 3 Nodal Planes
This orbital has zero overlap between adjacent p orbitals and therefore electrons in this orbital have the minimum possible delocalization. They are therefore the highest energy.
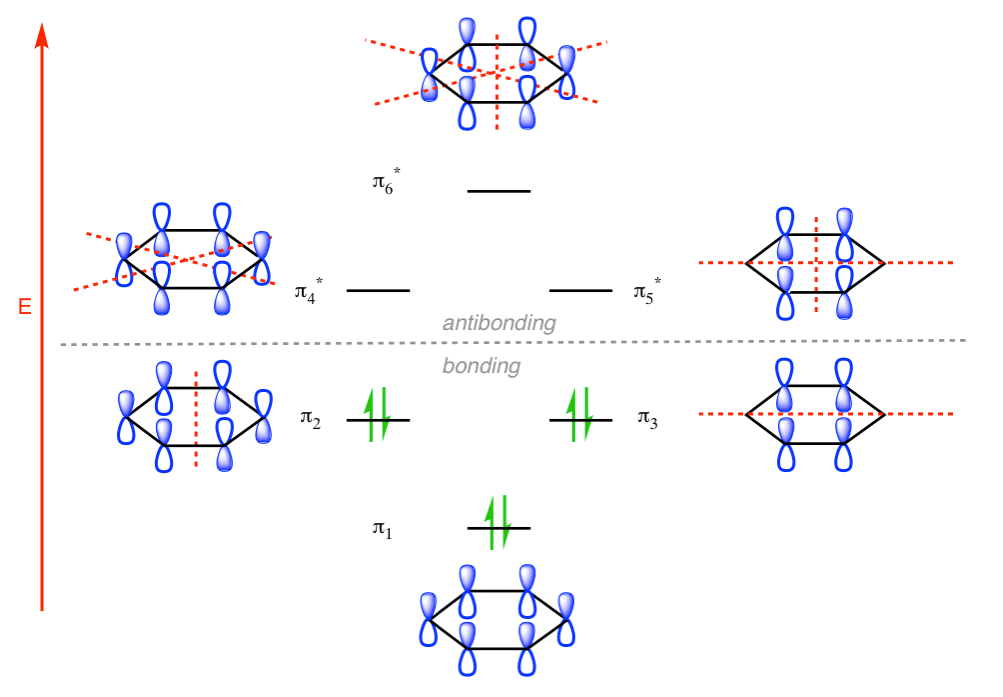
The pi molecular orbital of benzene
On attraction of positively charged metal cation and the negative charge on the non-bonding electrons of the ligand the interaction between a transition metal and ligands arises.
As a ligand approaches the metal ion, the electrons from the ligand will be closer to some of the d-orbitals and farther away from others, causing a loss of degeneracy. The electrons in the d-orbitals and those in the ligand repel each other due to repulsion between like charges. Thus the d-electrons closer to the ligands will have a higher energy than those further away which results in the d-orbitals splitting in energy.
The splitting can be affected by following factors:-
• Metal ion nature
• Metals oxidation state.
• Ligand arrangement around the metal ion
• Metal coordination number
Spectrochemical Series: The ability of ligands to cause a large splitting of the energy between the orbital is essentially independent of the metal ion and the spectrochemical series is a list of ligands ranked in order of their ability to cause large orbital separations.
I- < Br- < SCN- ~Cl- < F- < OH- ~ ONO- < C2O42- < H2O< NCS- < EDTA4-<NH3 ~ pyr ~ en < bipy < phen < CN- ~ CO
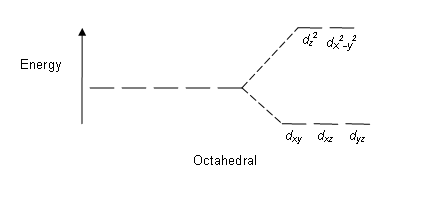
Octahedral: The crystal field stabilization energy (CFSE) is the stability that results from placing a transition metal ion in the crystal field generated by a set of ligands. It arises due to the fact that when the d orbitals are split in a ligand field, some of them become lower in energy than before. For example, in the case of an octahedron, the t2g set becomes lower in energy. As a result, if there are any electrons occupying these orbitals, the metal ion is more stable in the ligand field by the amount known as the CFSE. Conversely, the eg orbitals are higher in energy. So, putting electrons in them reduces the amount of CFSE.
Square Planar: Crystal field stabilization is applicable to the transition-metal complexes of all geometries. The reason that many d8 complexes are square-planar is the very large amount of crystal field stabilization that this geometry produces with this number of electrons.
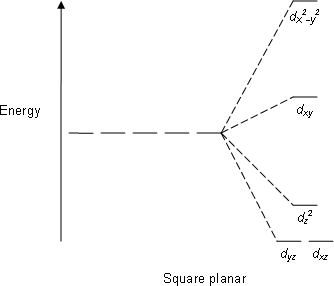
E.g; XeF4, PtCl2−
Tetrahedral:
dxy dxz dyz
The electron density (i.e., the lobes of the orbitals) lies nearest to the point charges.
dx2-y2 dz2
The electron density lies further away from the point charges.
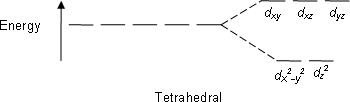
E.g; CH4
d-Orbital Splitting
CFT focuses on the interaction of the five (n − 1)d orbital’s with ligands arranged in a regular array around a transition-metal ion. Other common structures, such as square planar complexes, can be treated as a distortion of the octahedral model. According to CFT, an octahedral metal complex forms because of the electrostatic interaction of a positively charged metal ion with six negatively charged ligands or with the negative ends of dipoles associated with the six ligands. In addition, the ligands interact with one other electrostatically. According to VSEPR theory the lowest-energy arrangement of six identical negative charges is an octahedron, which minimizes repulsive interactions between the ligands.
The energies of the d-orbital of a transition-metal ion are affected by an octahedral arrangement of six negative charges. The five d-orbital are initially degenerate. On the distribution of six negative charges uniformly over the surface of a sphere, the d-orbital remain degenerate, but their energy will be higher due to repulsive electrostatic interactions between the spherical shell of negative charge and electrons in the d-orbital. Placing the six negative charges at the vertices of an octahedron does not change the average energy of the d-orbital, but it does remove their degeneracy: the five d-orbital split into two groups whose energies depend on their orientations. The dz2 and dx2−y2 orbital point directly at the six negative charges located on the x, y, and z axes. Consequently, the energy of an electron in these two orbital will be greater than it will be for a spherical distribution of negative charge because of increased electrostatic repulsions. In contrast, the other three d orbital (dxy, dxz, and dyz, collectively called the t2g orbitals) are all oriented at a 45° angle to the coordinate axes, so they point between the six negative charges. The energy of an electron in any of these three orbital is lower than the energy for a spherical distribution of negative charge.
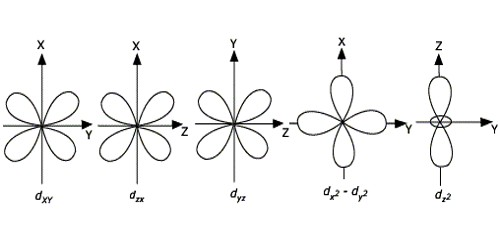
The Nature of the Ligands
The Δo observed for a series of complexes of the same metal ion depends strongly on the nature of the ligands. For a series of chemically similar ligands, the magnitude of Δo decreases as the size of the donor atom increases. E.g.: Δo values for halide complexes generally decrease in the order F− > Cl− > Br− > I−. In addition, a small neutral ligand with a highly localized lone pair, such as NH3, results in significantly larger Δo values that might be expected. Because of the lone pair points directly at the metal ion, the electron density along the M–L axis is greater than for a spherical anion such as F−. The experimentally observed order of the crystal field splitting energies produced by different ligands is called the spectro-chemical series, shown here in order of decreasing Δo:
CO≈CN−> NO−2>en>NH3>SCN−>H2O>oxalate2−>OH−>F>acetate−>Cl−>Br−>I−
Magnetic Property of transition elements:
A magnetic field is generated due to the orbital motion and spin of the electron. The spinning of an electron in an orbit is very much similar to flow of electric current in a closed circuit. Therefore, an unpaired electron is regarded as a micro magnet which has a definite magnetic moment. A substance which contains an unpaired electron when placed in a magnetic field interacts with the applied field. Consequently, an attractive force is exerted and the paramagnetic property is shown. The number of unpaired electrons determines the magnitude of magnetic moment. Higher the number of unpaired electrons more is the magnetic moment and greater will be the paramagnetic behavior of the substance.
In the case of paired electrons, the electrons in each pair will have opposite spin. The magnetic field created by the electrons of same pair is equal and opposite in nature. Hence, the magnetic field which is created by one electron is cancelled by the other. So the net effect of the magnetic moment is zero. These kinds of substances show diamagnetic property and are repelled by the applied magnetic field.
Q-1 Find the crystal field stabilization energy (CFSE) (in kJ/mol) for complex, [Ti(H2O)6]3+. According to CFT, the first absorption maximum is obtained at 20,3000 cm−1 for the transition.
ΔE=hcv−34
=6.63×10 Js × 3.00×108 m/s × 20300cm−1 × 1m100cm
=4.037×10−19J
This is energy change for one ion
For one mole
ΔE=4.037×10−19 J/ion × 6.02 × 1023 ions/mol
=243067 J/mol
But, 1kJ=1000J
∴ ΔE = 243 kJ/mol
But, T1 ion has d' electron configuration
CFSE= 0.4 ΔE
Q2 Calculate the octahedral crystal field splitting energy in kJ/mol for [Fe(CN)6] 4- , if the wavelength of the most intensely absorbed light is 305 nm.
Δo = hνν = c/λ Δo = hc/λ
Δo = 6.6261 x 10-34 Js (2.9979 x 108 m/s)/ 305 x 10-9 m
6.513 x 10-19 J x 1 kJ/1000J x 6.02214 x 1023 molecules /mol= 392 kJ/mol
Q-3 Calculate the crystal field stabilization energy (CFSE) for high spin Fe2+. Do not include pairing energy.
CFSE = 4(-2/5 Δo) + 2(+3/5 Δo)
= -2/5 Δo
Q4 Calculate the crystal field stabilization energy (CFSE) for low spin Fe2+. Do not include pairing energy.
CFSE = 6(-2/5 Δo) = -12/5 Δo




