Unit – 2
Equilibrium
Reversible reaction
Any changes which can be reversed or are a temporary conversion are known as reversible changes. The reactions which are reversible are called reversible reactions. In this reaction, one substance is modified into another form but a new compound is not formed. Processes such as melting, boiling, evaporation, freezing, condensation, dissolution are reversible changes. Few examples are melting of wax, freezing of ice, boiling water which evaporates as steam and condenses back to water.
Reactions are an interaction of two or more compounds called reactants to produce a product(s). In a reversible reaction, reactants and products formed are connected by a two-way arrow (⇌). This means reactants can be obtained back from the products.
Consider the reaction below,
A +B ⇌ C + D
Here, A and B are two reactants which react to give C and D. The two-headed arrow indicates that reaction is reversible and the reactants, A and B can be obtained from C and D.
Irreversible reaction
In contrast to reversible reaction, irreversible changes are permanent changes. Reactants react to form an entirely new compound and cannot be reversed. Heating, burning, mixing, powdering are few processes which cause irreversible changes. A common observable example is the cooking of raw egg which can’t be converted back to its original form. Ash obtained by the combustion of paper or any other substances is another example.
When a reaction is taking place in a unidirectional way such reactions are called irreversible reactions. In such reactions in a period of time reactants react completely to form a product. Here reaction is denoted by a one-way arrow (→).
For example,
A → B +C
Here, A is the reactant which is completely converted into products B and C which do not react to form A.
DEFINITION OF CHEMICAL CHANGE - DEFINITION
A change in which one or more new substances are formed is called a chemical change. A chemical change is also called a chemical reaction.
Examples are burning of any substance, rusting of iron.
REVERSIBLE REACTION - DEFINITION
A reversible reaction is a chemical reaction that can proceed in both forward and backward direction.
aA+bB⇋cC+dD
A and B can react to form C and D or, in the reverse reaction, C and D can react to form A and B.
IRREVERSIBLE REACTIONS - DEFINITION
If a reaction cannot take place in the reverse direction, i.e., the products formed do not react to give back the reactants under the same condition is called an irreversible reaction.
BaCl2(aq)+Na2SO4(aq)→BaSO4(s)+NaCl(aq)
Statement of law of mass action and its kinetic derivation
The law of mass action states that the rate of a reaction is proportional to the product of the concentrations of each reactant.
The law of mass action is used to explain the performance shown by solutions in dynamic equilibria. The law of mass action also implies that at a state of chemical equilibrium, the ratio of the reactant concentration and the ratio of the product concentration is constant.
The Equilibrium Constant (Kc)
For a given temperature, the concentration of the reactants and products are constant at equilibrium. Considering the simple reaction that is reversible where A & B are the reactants whereas C & D are the products.
A + B ⇌ C + D
A mixture of reactant and products at a state of equilibrium is called as equilibrium mixture. There is a relationship that exists between the concentration of reactants and the concentration of products for a given equilibrium mixture. This relation can be equated as follows.
 Kc = [C][D]
Kc = [C][D]
[A][B]
Here, Kc is called the equilibrium constant. In this equation, the concentration of A at equilibrium is represented as [A] (similarly for B, C, and D), and the stoichiometric coefficients of the reactants and products are 1. It has been experimentally observed that the equilibrium constant is also dependant on the stoichiometric coefficients of the reactants and products.
Therefore, the law of mass action dictates that the equilibrium constant, at a given constant temperature, is equal to the product of the concentration of products raised to the respective stoichiometric coefficients divided by the product of the reactant concentrations, each raised to the corresponding stoichiometric coefficient.
This is also known as the equilibrium law or the law of chemical equilibrium
Equilibrium constant for homogeneous and heterogeneous reaction
Introduction to Homogeneous equilibrium
In order to simplify the problems and understand the concept, we divide such reactions into different categories, namely, the homogeneous reactions, where the components involved in the reaction are present in the same phase and the heterogeneous reactions, where the components involved are present in different phases. The methods of dealing with both the reactions are different and so is the determination of the equilibrium state. In this section, we will learn about a homogeneous equilibrium and the calculation of equilibrium constant for a homogeneous reaction.
Equilibrium Constant for Homogeneous Reaction
Let us consider a homogeneous system, given by the following reaction
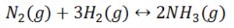
Here, we see that gaseous nitrogen reacts with gaseous hydrogen to form gaseous ammonia. Let us now calculate the equilibrium constant for the above reaction.
In terms of the molar concentration of the components in the reaction, we can write
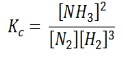
As we know, for reactions involving gases, we express the equilibrium concentration in terms of the partial pressure.
Using the ideal gas equation,
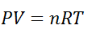
Where P is the pressure of the system, V is the volume of the system, n is the number of moles of components present in the system, R is the universal gas constant and T is the temperature of the system.
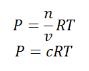
Here, c is the concentration of the system. We can also write it as,
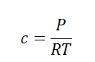
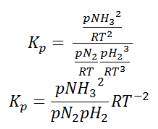
So, equilibrium constant can also be written as,
Here, we note that the general expression for Kc and Kp can be written as,
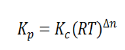
Where Δn is defined as the number of moles of gaseous products – the number of moles of gaseous reactants.
Equilibrium Constant for Heterogeneous Reaction
When the state of equilibrium in a system has components in more than one phase it is termed as a heterogeneous equilibrium. For example, if we take a container with ice and water at a temperature that is allowing the existence of both the phases simultaneously, such that, both ice and water are present in a state of equilibrium. This state is termed as heterogeneous equilibrium.
In terms of its equation, this can be written as:
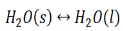
In another example, we can consider an aqueous solution of a solid such as calcium hydroxide. We notice that the solid calcium hydroxide is in equilibrium with its saturated solution.
Writing the equilibrium constant for heterogeneous reactions is different from that of the homogeneous reactions. For example, consider the thermal dissociation of calcium carbonate into calcium oxide and carbon dioxide. Here, we can see that the equilibrium constant for the dissociation of the reactant into its products is only dependent on the gaseous component, as the solid and the liquid reactants are considered to be constant.
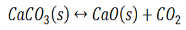
Here, the components CaCO3 and CaO are solids, so their molar concentration remains constant throughout the reaction. Therefore, the equilibrium constant can be written as,
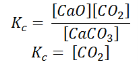
Also, in terms of Kp, we can write
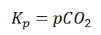
Where p denotes the partial pressure. In other words, we can state that, at a given temperature, there is a constant concentration or partial pressure of CO2 in the equilibrium reaction with CaO and CaCO3.
Representation of the Equilibrium Constant
For a balanced reaction of the type,
aA + bB ⇌ cC + dD
According to the law of mass action, the constant value obtained by relating equilibrium concentrations of reactants and products is called the equilibrium constant. For the forward reaction, this is given by
 Kc = [C]c[D]d
Kc = [C]c[D]d
[A]a[B]b
The equilibrium constant for the reverse reaction is the inverse of the forward reaction and is given by:

 K′c = 1 = [A]a[B]b
K′c = 1 = [A]a[B]b
Kc [C]c[D]d
If the coefficients of the chemical equation are multiplied by a factor ‘n’ then the equilibrium constant is raised by the power ‘n’ i.e., the constant becomes Knc.
Equilibrium Constant Representation | Expressed in terms of | Expressed as |
Kc | Concentrations of reactants and products |
[A]a[B]b |
Kp | Partial pressures of reactants and products. (Only for the substances which are in gaseous state) |
paApbB |
Kx | Mole fractions of reactants and products |
[XA]a[XB]b |



Relation between Kc, Kp and Kx
Kp = Kc (RT)Δng
Kx = Kp (RT)Δng
Where,
Δng = moles of gaseous products – moles of gaseous reactants.
Kc is the equilibrium constant expressed in terms of the concentration of the reactants/products. Similarly, Kp is the constant in terms of the partial pressures of the substances and K x is expressed in terms of the mole fraction.
Le Chatelier’s Principle
Le Chatelier’s principles, also known as the equilibrium law, are used to predict the effect of some changes on a system in chemical equilibrium (such as the change in temperature or pressure). The principle is named after the French chemist Henry Louis Le Chatelier.
Le Chatelier said that equilibrium adjusts the forward and backward reactions in such a way to accept the changes affecting the equilibrium conditions.
Le Chatelier's principle can be used to predict the behavior of a system due to changes in pressure, temperature, or concentration. Le Chatelier's principle implies that the addition of heat to a reaction will favor the endothermic direction of a reaction as this reduces the amount of heat produced in the system.
Effect of Concentration Changes on Equilibrium and Product Formation
As per Le Chatelier’s principles, the only way of equilibrium to accept more reactant is to increase product formation. The forward reaction is favoured when the concentration of the reactant is increased. The equilibrium of the reaction shift towards the use of reactants in the reaction, which decreases the concentration of the reactants.
Similarly, the addition of product (concentration/pressure) shall increase the backward reaction to decrease the product concentration. The backward reaction is favoured when the concentration of the reactant decreases and equilibrium of the reaction shift towards the production of reactants and concentration of the reactants will be more.
Effect of Change of Volume, Pressure, or Inert Gas on Equilibrium and Product Formation
Kp = Kc (RT)Δn = Kc (p/v) Δn
Change of volume, pressure, or inert gases has no effect on reactions of liquids and solids. They may have an effect in gaseous reactions and that too only when the difference in the sum of the number of reactant and product molecules (∆n) is not zero.
When ∆n = 0:
As per the Le Chatelier’s principles, there will be no effect on Equilibrium and Product Formation on changing the volume, pressure or inert gas.
When ∆n = +ve:
Increase of pressure or decrease in volume will decrease the formation of the product. Decrease of pressure or increase of volume shall have the opposite effect of increasing the product formation.
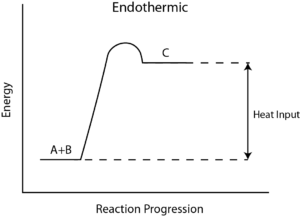
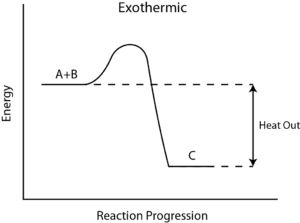
Effect of Change of Temperature on Equilibrium and Product Formation
The individual reaction in the equilibrium can be either endothermic or exothermic. Likewise, at equilibrium net energy involved may make the reversible reactions either endothermic or exothermic.
According to Le Chatelier’s Principles,
Application of Le Chatelier’s
Heats of chemical reactions
The Heat of Reaction (also known and Enthalpy of Reaction), occurs in the constant pressure, it is the change in the enthalpy of a chemical reaction. Enthalpy is a state function and a thermodynamic unit for measurement. The enthalpy is used to calculate the amount of energy per mole that is either produced or released in the reaction. Since enthalpy is derived from pressure, volume, and internal energy, all of which are state functions.
Introduction
ΔHΔH, or the change in enthalpy is used as a unit of measurement to calculate the change in energy of a system, when there are difficulties in calculating the change in internal energy of the system, by simultaneously measure the amount of heat and work exchanged. Given a constant pressure, the change in enthalpy can be measured as
ΔH=q (1)(1)ΔH=q
The notation ΔHº or ΔHºrxn then arises to explain the precise temperature and pressure of the heat of reaction ΔH. The standard enthalpy of reaction is symbolized by ΔHº or ΔHºrxn and can take on both positive and negative values. The units for ΔHº are kiloJoules per mole, or kj/mol.
ΔH and ΔHºrxn
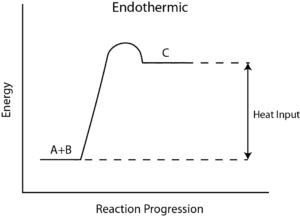
- a positive value indicates the products have greater enthalpy, or that it is an endothermic reaction (heat is required)
- a negative value indicates the reactants have greater enthalpy, or that it is an exothermic reaction (heat is produced)
The Standard State: The standard state of a solid or liquid is the pure substance at a pressure of 1 bar (105 Pa) and at a relevant temperature.
The ΔHºrxn is the standard enthalpy of a reaction or the standard enthalpy of a reaction, and like ΔH also measures the enthalpy of a reaction. However, ΔHºrxn takes place under "standard" conditions, the standard conditions take place at 25º C and 1 atm. The benefit of a measuring ΔH under standard conditions lies in the ability to relate one value of ΔHº to another, since they occur under the same conditions.
How to Calculate ΔH Experimentally
Enthalpy can be measured experimentally through the use of a calorimeter. A calorimeter is an isolated system which has a constant pressure, so ΔH=q=cpsp x m x (ΔT)
How to calculate ΔH Numerically
To calculate the standard enthalpy of reaction the standard enthalpy of formation must be utilized. Another, more detailed, form of the standard enthalpy of reaction includes the use of the standard enthalpy of formation ΔHºf:
ΔH⊖=∑ΔvpΔH⊖f(products)−∑ΔvrΔH⊖f(reactants) (2)
with
Since enthalpy is a state function, the heat of reaction depends only on the final and initial states, not on the path that the reaction takes. For example, the reaction A→BA→B goes through intermediate steps (i.e., C→DC→D), but A and B remain intact.
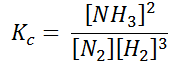
Therefore, one can measure the enthalpy of reaction as the sum of the ΔH of the three reactions by applying Hess’ Law
Since the ΔHº represents the total energy exchange in the reaction this value can be either positive or negative.
Example 11: the combustion of acetylene
Calculate the enthalpy change for the combustion of acetylene (C2H2C2H2)
Solution
1) The first step is to make sure that the equation is balanced and correct. Remember, the combustion of a hydrocarbon requires oxygen and results in the production of carbon dioxide and water.
2C2H2(g)+5O2(g)⟶4CO2(g)+2H2O(g)(3)(3)2C2H2(g)+5O2(g)⟶4CO2(g)+2H2O(g)
2) Next, locate a table of Standard Enthalpies of Formation to look up the values for the components of the reaction.
3) First find the enthalpies of the products:
ΔHºf CO2 = -393.5 kJ/mole
Multiply this value by the stoichiometric coefficient, which in this case is equal to 4 moles.
vpΔHºf CO2 = 4 mol (-393.5 kJ/mole)
= -1574 kJ
ΔHºf H2O = -241.8 kJ/mole
The stoichiometric coefficient of this compound is equal to 2 moles. So,
vpΔHºf H2O = 2 mol ( -241.8 kJ/mole)
= -483.6 kJ
Now add these two values in order to get the sum of the products
Sum of products (Σ vpΔHºf(products)) = (-1574 kJ) + (-483.6 kJ) = -2057.6 kJ
Now, find the enthalpies of the reactants:
ΔHºf C2H2 = +227 kJ/mole
Multiply this value by the stoichiometric coefficient, which in this case is equal to 2 moles.
vpΔHºf C2H2 = 2 mol (+227 kJ/mole)
= +454 kJ
ΔHºf O2 = 0.00 kJ/mole
The stoichiometric coefficient of this compound is equal to 5 mole. So,
vpΔHºf O2 = 5 mol (0.00 kJ/mole)
= 0.00 kJ
Add these two values in order to get the sum of the reactants
Sum of reactants (Δ vrΔHºf(reactants)) = (+454 kJ) + (0.00 kJ) = +454 kJ
The sum of the reactants and products can now be inserted into the formula:
ΔHº = Δ vpΔHºf(products) -? vrΔHºf(reactants)
= -2057.6 kJ - +454 kJ
Reaction enthalpy
During any chemical reaction, heat is either absorbed or given out. The heat exchange between the chemical reaction and its environment is reaction enthalpy (H). This cannot be measured directly. For this, there is a measurement of change in the temperature of a reaction over time to the final change in enthalpy denoted by ΔH.
From the value of ΔH, whether in a reaction heat is absorbed from the environment [endothermic] or is given off to the environment [exothermic] is determined. In general, ΔH is given as, ΔH = m×s×ΔT, where m is the mass of reactants, s is the specific heat of products, and ΔT is a change in temperature during the completion of the reaction.
How to Calculate the Reaction Enthalpy?
To find enthalpy of a chemical reaction we need to follow the below mentioned steps.
Finally, we will get the enthalpy of the chemical reaction.
Example of Enthalpy Calculation
Hydrogen reacts with oxygen to give water. The chemical reaction is,
2H2 + O2 → 2H2O
Reactants Products
2H2 + O2 = 2H2O
2×[2g] 1×[32g] = 4g + 32g =36g
2H2 + O2 = H2O
The specific heat of water is 4.2 joule/gram°C
ΔT = T1 – T2 (T1 = 185K and T2 = 95 K) = 185K – 95K
ΔT = -90K
In the above reaction, the reactants had more temperature, and after the completion of the reaction, the product got cooled off.
ΔH = 36g×42 jg-1k-1 × (-90) K = -13608 joule
ΔH < 0
This makes logical sense since hot gases hydrogen (H2) and oxygen(O2) react together and let off heat to the environment. Then water [H2O] forms, indicating that it is an exothermic reaction.
Which other methods are used to calculate enthalpy?
Here is the alternate method
H2 + F2 → 2HF
We need bond energy to break H2 and F2 molecules of 436kJ/mol and 158kL/mol, respectively. The bond energy required to form HF molecule is -568kJ/mol.
H2 + F2 → 2HF
436kJ/mol 158kJ/mol 2*(-568)
= -1136 kJ/mol
Adding these values together we get,
436 + 158 + (-1136) = -542kJ/mol
Standard enthalpy changes
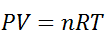
Enthalpy change: Standard Enthalpy of Reaction
While dealing with a chemical reaction, the knowledge of enthalpy and standard enthalpy is very important. It helps in calculating the temperature and pressure required for any chemical reaction. It is also required for calculating the amount of heating and cooling necessary when the reaction is carried out commercially (at a large scale), for example, for the mass production of any component such as ammonia, calcium carbonate, oxygen etc.
Enthalpy change
Enthalpy change is the standard enthalpy of formation, which has been determined for a vast number of substances. In any general chemical reaction, the reactants undergo chemical changes and combine to give products. It can be represented by the following equation:
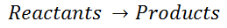
For any such reaction, the change in enthalpy is represented as ΔrH and is termed as the reaction enthalpy. The reaction enthalpy is calculated by subtracting the sum of enthalpies of all the reactants from that of the products.
Mathematically,
ΔtH = Sum of enthalpies of the product – Sum of the enthalpies of the reactants.
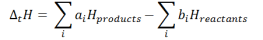
Here, the constants ai and bi denotes the stoichiometric coefficients of the products and the reactants respectively for the balanced chemical reaction under consideration.
Standard enthalpy of reaction
Any Enthalpy of a reaction is dependent on its physical conditions of the surrounding such as pressure, temperature etc. In order to specify the standard enthalpy of any reaction, the standard enthalpy of a reaction is calculated when the components that participate in the reaction are in standard form, the components include reactants and products. Therefore, the standard enthalpy of reaction is the enthalpy change that occurs in a system when a matter is transformed by a chemical reaction under standard conditions.
As per convention, the standard state for any substance at a specified temperature is its pure form at a pressure of 1 bar. For example, liquid ethanol at 298 K and 1 bar of pressure is said to be in its standard state in its pure form. It is important to note that the data for the standard state for a substance is taken at 298K. The standard enthalpy of a reaction is denoted as ΔrHs. At constant pressure, the heat of the reaction is exactly equal to the enthalpy change, of the reacting system.
Hess’s Law
Introduction
Hess's Law is named after Russian Chemist and Doctor Germain Hess. He was successful in formulating the early principles of thermochemistry. His law on chemistry was published in 1840 in one of his famous papers. Hess’s law was introduced due to enthalpy being a state function, that helps us to calculate the overall change in enthalpy by simple addition of the changes that take place in each step of the reaction till the products are formed. All the individual equations must be balanced and all steps need to proceed at the same temperature. The principle underlying Hess's law does not just apply to Enthalpy and can be used to calculate other state functions like changes in Gibb’s Energy and Entropy.
Definition: Hess's Law
The heat of any reaction ΔH∘fΔHf° for a specific reaction is equal to the sum of the heats of reaction for any set of reactions which in sum are equivalent to the overall reaction:
(Although we have not considered the restriction, applicability of this law requires that all reactions considered proceed under similar conditions: we will consider all reactions to occur at constant pressure.)
Why it works
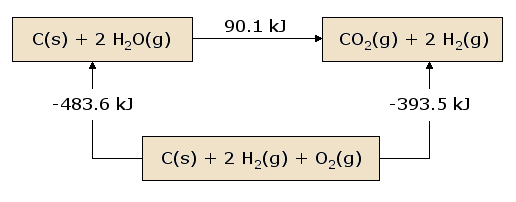
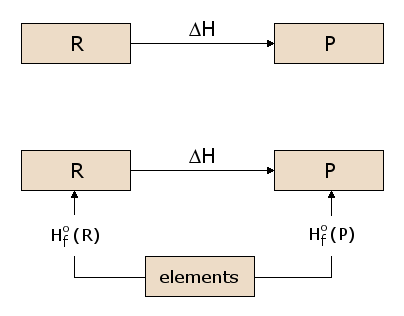
A pictorial view of Hess's Law as applied to the heat of equation. In figure 1, the reactants C(s) + 2 H2O(g) are located in a box that represent the materials that are involved in the reaction prior to the reaction.
The products CO2(g) + 2 H2(g) are located together in the second box that represent the state the materials that are involved in a reaction., the arrows that connect these two boxes represent the heat of the reaction.
Now the same materials are placed in the third box, that contains C(s), O2(g), and 2 H2(g). the box is connected to the other boxes with reaction arrows labelled by heat of reactions.
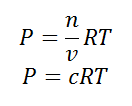
Pictorial View of Hess's Law.
This picture of Hess's Law reveals that the heat of reaction along the "path" directly connecting the reactant state to the product state is exactly equal to the total heat of reaction along the alternative "path" connecting reactants to products via the intermediate state containing C(s)C(s), O2(g)O2(g), and 2 H2(g)H2(g). The net heat that is absorbed or evolved during a reaction, is independent to the path that connects the reactants and products. (This statement is again subject to our restriction that all reactions in the alternative path must occur under constant pressure conditions).
As we begin from the reactant box and go through the complete circuit of other boxes, and leading back to the reactant box, when we consider the net heat of the reaction. We discover that the net heat transferred (again provided that all reactions occur under constant pressure) is exactly zero. This is a state of conservation of energy.: the energy in the reactant state does not depend upon the processes which produced that state.
By perception energy function is the value for the reactants and is independent of how the reactant state is prepared. Similarly, the value of this energy functioning the product state is independent of how the products are prepared. We choose this function, H, so that the change in the function, ΔH = Hproducts - Hreactants, is equal to the heat of reaction q under constant pressure conditions. H, which we call the enthalpy, is a state function, since its value depends only on the state of the materials under consideration, that is, the temperature, pressure and composition of these materials.
For calculating the heats of a reaction, the energy state function H is considered. The figure given below the reactants R being converted to products P. ΔH is a state function that is used to calculate the heat absorbed or released in the reaction. When R and P are used to calculate ΔH along the path, we should consider the two reaction that go through an intermediate state that consists of all the atoms that are involved in a reaction and each in its elemental form. This intermediate stage can be used for all chemical reactions.in the initial figure given above the reaction are C, H, and O, each of which are represented in the intermediate state in elemental form. But in subfigure given below is the ΔH for the overall reaction is now the difference between the ΔH in the formation of the products P from the elements and the ΔH in the formation of the reactants R from the elements.
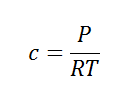
Figure: Calculation of ΔH.
The ΔH values for formation of each material from the elements are thus of general utility in calculating ΔH for any reaction of interest. We therefore define the standard formation reaction for reactant R, as
elements in standard state 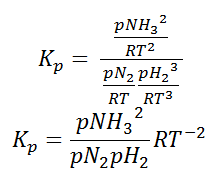 R
R
and the heat involved in this reaction is the standard enthalpy of formation, designated by ΔHf°. The subscript f, standing for "formation," indicates that the ΔH is for the reaction creating the material from the elements in standard state. The superscript ° indicates that the reactions occur under constant standard pressure conditions of 1 atm. The heat of any reaction can be calculated from
ΔH∘f=ΔH∘f, products−ΔH∘f, reactants
ΔHf°=ΔHf, products°−ΔHf, reactants°
The enthalpy of a reaction is not dependant on the elementary steps, but on the initial state of the reactants and the final state of products. This means that the enthalpy of the reaction scales proportionally to the moles used in the reaction.
Since enthalpy is a state function, it is path independent. Therefore, it does not matter what reactions one uses to obtain the final reaction.
Kirchhoff’s Law
Kirchhoff’s First Law – The Current Law, (KCL)
Kirchhoff’s Current Law or KCL, states that the “the total amount of current or charge that enters a junction or a node is exactly equal to the charge that leaves the node, no charge is lost within the node, as the charge has no place to go but to leave the circuit. “. In other words, the algebraic sum of ALL the currents entering and leaving a node must be equal to zero
Kirchhoff’s Second Law – The Voltage Law, (KVL)
Kirchhoff’s Voltage Law or KVL, states that “in any closed loop network, the total voltage around the loop is equal to the sum of all the voltage drops within the same loop” which is also equal to zero. In other words, the algebraic sum of all voltages within the loop must be equal to zero
Components are said to be connected together in Series if the same current value flows through all the components.
Components are said to be connected together in Parallel if they have the same voltage applied across them.
Relationship between Enthalpy (H) and Internal energy (U)
The change in the Internal energy of a system is the sum of the heat transferred and the work done. At constant pressure, heat flow (q) and Internal energy (U) are related to the system's enthalpy (H). The heat flow is equal to the change in the Internal energy of the system plus the PV work done
Enthalpy a process or activity takes place at constant pressure, the heat released or absorbed is equal to the Enthalpy change. Enthalpy is occasionally called or known as “heat content”, “enthalpy” is derived from the Greek word which means “warming”. Enthalpy(H) is the sum of the internal energy(U) and the result of pressure(P) and volume(V).
Enthalpy H is written as,
H = U + PVm
Where, H = is the Enthalpy of the system
U = is the Internal energy of the system and is entirely dependent on the state functions T, p and U.
Let Hi be the enthalpy of a system in the initial state and Hf be the enthalpy of a system in the final state. Let Ui and Vi be the internal energy and volume in the initial state and Uf and Vf be the internal energy and volume in a final state.
Now, as we know that,
H=U+PV
Therefore,
Hi=Ui+PVi......(1)
Hf=Uf+PVf......(2)
Subtracting equation (1) from (2), we have
Hf−Hi=(Uf+PVf) −(Ui+PVf)
⇒Hf−Hi=(Uf−Ui) +P(Vf−Vi)
⇒ΔH=ΔU+PΔV..... (3)
Here, ΔU and PΔV are the change in internal energy and work energy respectively.
Hence equation (3) is the relationship between H and U
Enthalpy can also be written as
ΔH=ΔU+ΔPV.
P = is the Pressure of the system
V = Volume of the system Enthalpy is not directly measured, but the change in enthalpy (ΔH) is measured, which is the heat lost or added by the system,
Bond Energy
Bond energy and its determination
The Bond energy is determined by calculating the heat that is required to break one mole of molecules into their individual atoms, it denotes the average energy that is associated with breaking the individual bonds of a molecule. The higher the bond energy, the ‘stronger’ we say the bond is between the two atoms, and the distance between them (bond length) is smaller.
For instance, the HO-H bond in a molecule of water involves 493kJ/mol to break and generate the hydroxide ion (OH–). Further the breaking of the O-H bond in the hydroxide ion needs an additional of 424 kJ/mol. Therefore, the bond energy of the covalent O-H bonds in water is determined to be the average of the both the values, or 458.9 kJ/mol. These energy values (493 and 424 kJ/mol) are required to break successive O-H bonds in a water molecule and are generally called as ‘bond dissociation energies,’ and they are different from the bond energy. Therefore, the bond energy is the average of the bond dissociation energies in a molecule.
The exact properties of a specific kind of bond are determined in part by the nature of the other bonds in the molecule; for example, the energy and length of the C–H bond will vary depending on what other atoms are bonded to the carbon atom. Similarly, the C-H bond length can vary by as much as 4% between different molecules. For this reason, the values of bond energy and bond length are usually averages taken over a variety of compounds that contain a specific atom pair.
We can apply bond energy values to determine the enthalpy of a compound’s formation.
Enthalpies of ions in Solutions.
Enthalpy changes of solution
The enthalpy change with respect to a solution is the enthalpy change where 1 mole of an ionic substance dissolves in water to form a solution of indefinite dilution. The enthalpy of a solution is either negative or positive, in other words, some ionic substances dissolved endothermically (for example, NaCl); others dissolve exothermically (for example NaOH).
An infinitely solution that is dilute in one where there is an infinitely dilute solution is one where there is an adequately excess amount of water such that adding any further does not involve any heat to be absorbed or evolved. Therefore when 1 mole of sodium chloride crystals are dissolved in an excess of water, the enthalpy change of solution is found to be +3.9 kJ mol-1. The change is slightly endothermic, and so the temperature of the solution will be slightly lower than that of the original water.
When a substance dissolves why is it that heat is sometimes dissolved or absorbed. An imaginary process can be put forward the crystal lattice is first broken up into its separate gaseous ions and then those ions have water molecules wrapped around them. That is how they exist in the final solution.
Factors affecting the size of hydration enthalpy
Hydration enthalpy is a measure of the energy released when attractions are set up between positive or negative ions and water molecules.
The size of the hydration enthalpy is governed by the amount of attraction between the ions and the water molecules.
Estimating enthalpies of solution from lattice enthalpies and hydration enthalpies
The hydration enthalpies for calcium and chloride ions are given by the equations:
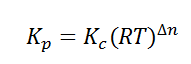
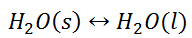
The following cycle is for calcium chloride, and includes a lattice dissociation enthalpy of +2258 kJ mol-1. We have to use double the hydration enthalpy of the chloride ion because we are hydrating 2 moles of chloride ions. Make sure you understand exactly how the cycle works.
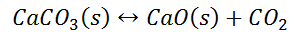
So
ΔHsol = +2258 - 1650 + 2(-364)
ΔHsol = -120 kJ mol-1
As the case above the negative hydration enthalpies are found to be more than the positive lattice dissociation enthalpy, depending on the size of the lattice enthalpy and the hydration enthalpy the enthalpy of solution corresponds to being positive or negative.
Thermodynamics terms:
Thermodynamic systems:
Thermodynamics in biology is the study of energy transfers that take place in molecules or in a collection of molecules. Generally, when dealing with Thermodynamics, a specific item or a collection of items that we are dealing with is called a system, while everything that's not included in the system, we’ve defined is called the surroundings.
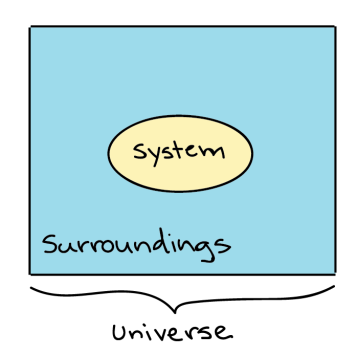
Generalized depiction of the system (a circle), the surroundings (a square surrounding the circle), and the universe (system + surroundings).
For example, if a pot of water is heated on the stove, the system included the stove, pot and water, while the surrounding could be the rest of the items in the kitchen, the decision of what to define as the system is arbitrary (up to the observer), and depending on what you wanted to study, we could equally well make just the water, or the entire house, part of the system. The system and the surroundings together make up the universe.
There are three types of systems in thermodynamics: open, closed, and isolated.
we, like other organisms, are an open system. We are constantly exchanging matter and energy with our surroundings for e.g. when we eat a carrot or lift a bag of laundry onto a table, or a simple breathing. In each case, you are exchanging energy and matter with your environment.
Intensive and Extensive properties
Thermodynamic properties can be divided into 2 (two) general classes such as intensive and extensive properties.
An intensive property, is the system’s physical property that does not depend on the size or the amount of material in the system. In contrast the extensive property of a system is not dependent on the system size or the amount of the material. According to the definitions, density, pressure and temperature are intensive properties and volume, internal energy are extensive properties.
Symbols for representing properties: Extensive properties are symbolized by upper case (capital) letter such as V (volume), KE (kinetic energy), PE (potential energy), etc. Intensive properties are symbolized by lower case letters such as v (specific volume), ke (specific kinetic energy), e, u (specific internal energy), h (specific enthalpy), etc. Mole based properties are symbolized by lower case letters with overbars. For example, kinetic energy and molar specific potential energy respectively.
Exceptions: Temperature (intensive), mass (extensive), and number of moles (extensive). The use of symbols for temperature, mass and moles are traditional.
Exceptions:
Symbols for Exceptional Properties
Parameter | Property | Traditional symbol |
Temperature | Intensive property | T |
Mass | Extensive property | M |
Number of moles | Extensive property | N |
Specific extensive properties, i.e., extensive properties per unit mass are intensive properties. For example: specific volume, specific energy, density, etc.
Thermodynamic Process
The four types of thermodynamic process are isobaric, isochoric, isothermal and adiabatic.
The Four Types of Thermodynamic Processes
The state of a given thermodynamics system can be expressed by various parameters like Pressure (P), temperature(T), volume(V) and internal energy(U), if ant two parameters are fixed, suppose pressure (P) and volume (V) of a fixed mass of gas, then the temperature (T) of the gas will be automatically fixed according to the equation PV=RT. No change can be made to T without altering P and V.
Work in Thermodynamic Processes
When the volume (V) of a system alters, it is said that pressure-volume work has occurred. A thermodynamic process occurring in a closed system in such a way that the rate of volume change is slow enough for the pressure (P) to remain constant and uniform throughout the system, is a quasi-static process. In this case, work (W) is represented as:
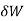 = PdV where δW is the infinitesimal work increment by the system ans dV is the infinitesimal volume increment.
= PdV where δW is the infinitesimal work increment by the system ans dV is the infinitesimal volume increment.
Also, W = 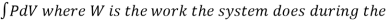 entire reversible process.
entire reversible process.
Isobaric Process
An isobaric process is one where the pressure of the system (often a gas) stays constant. 'Iso' means the same, and 'baric' means pressure. Pressure is related to the amount of force that the molecules apply to the walls of the container. Imagine that you have a gas inside a movable piston and you heat that gas up. By heating the gas up, you make the molecules move faster, which would normally increase the pressure. But at the same time the piston expands, increasing the volume and giving the molecules more room to move. Since the walls of the container are now bigger, the pressure can stay the same even though the molecules are moving faster. That makes it an isobaric process.
Since the pressure (P) is constant in this process, the volume of the system changes. The work (W) done can be calculated as W=P (Vfinal – V initial)
If ΔV is positive (expansion), the work done is positive. For negative ΔV (contraction) the work done is negative.
Isochoric Process
An isochoric process is one where the volume of the system stays constant. Again, 'iso' means the same and 'choric' means volume. Volume is the amount of space the material takes up. So, this would be like heating a gas in a solid, non-expandable container. The molecules would move faster and the pressure would increase, but the size of the container stays the same.
Isochoric Process
The volume remains constant in an isochoric process. Therefore, the system does not do any work (since ΔU=0, PΔV or W is also zero). Such a process in which there is no change in volume can be achieved by placing a thermodynamic system in a closed container which neither contracts nor expands. Thus, from the first law of thermodynamics (Q=ΔU + W), change in internal energy becomes equal to the heat transferred (ΔU = Q) for an isochoric process.
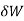 = PdV where δW is the infinitesimal work increment by the system ans dV is the infinitesimal volume increment.
= PdV where δW is the infinitesimal work increment by the system ans dV is the infinitesimal volume increment.
Also, W = 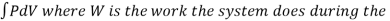 entire reversible process.
entire reversible process.
Isothermal Process
An isothermal process is one where the temperature of the system stays constant. Thermal relates to heat, which is in turn related to temperature. Temperature is the average heat (movement) energy of the molecules in a substance.
An example of an isothermal process would be if we took a gas held behind a movable piston and compressed that piston: the volume has decreased, and the pressure behind the piston has increased, since the molecules have less space in which to move. When you compress a piston, you're using energy - you're doing work on the gas - so normally the molecules would gain energy and move faster, and the temperature would increase. The temperature of the system remains constant in an isothermal process.
W = 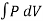
From Gas law
PV = nRT
P = nRT/V . Using the value of P in the work equation
W = nRT 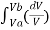
W = nRT ln(VB/VA)
If VB is higher than VA the work done will be positive or else negative
Since internal energy is temperature -dependent, ΔU=0 because the temperature is constant and thus from the first law of thermodynamics (Q= ΔU+W), we will get Q=W
Adiabatic process
The thermodynamic process in which there is no exchange of heat from the system to its surrounding neither during expansion nor during compression.
The adiabatic process can be either reversible or irreversible. Following are the essential conditions for the adiabatic process to take place:
No heat is exchanged with the system in an Adiabatic process (Q=0), its mathematical representation is
PV 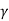 = K ( constant)
= K ( constant)
Also W = 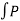 dV substituting the value of P in the work equation
dV substituting the value of P in the work equation
W = K 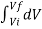 / V
/ V 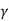
W = K [ (Vf 1-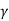 – Vi 1-
– Vi 1-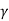 ) / 1 –
) / 1 – 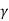 ]
]
Since Q=0 for an adiabatic process, from the first law of thermodynamics (Q=ΔU+W), we will get ΔU= -W. thus the internal energy will increase if the work done is negative and vice versa.
Path and process
An operation in which one or more of the properties of a system changes is called a change of state. The succession of states passed through during a change of state is called the path of the change of state.
The path of thermodynamic states that a system passes through as it goes from an initial state to a final state is known as the thermodynamic process.
Different thermodynamic processes are given in Table
Table: Different Thermodynamic Processes
Sr No | Name of the process | Parameter Held Constant | Remarks |
1 | Constant pressure (Isobaric) | p = constant | v = (mR/P) T |
2 | Constant volume(isochoric) process | V = constant | p = (mR/V) T |
3 | Constant temperature (isothermal) process | T = constant | pV = constant |
4 | Polytropic process | N | pVn = constant |
5 | Adiabatic process | no heat flow across the system boundary | pvγ = constant |
6 | Isoenthapic process | h = constant | h = constant |
State functions and Exact differentials
In thermodynamics it is very important to differentiate between exact and inexact differentials. The common terms in thermodynamics variables such as the internal energy (UU), volume, pressure, and temperature, and you probably heard about entropy (SS) and free energy (GG).
All these variables are used to specify the state of a system. These properties are not dependant on the way the system works but is used to specify the properties of the current state of the system. For example, if a system consists of 1 mol of Heat 298 K and 1 atm, the system regardless of its history will have variable such as internal energy, entropy, free energy. If the system is compressed at 2atm or is heated in gas from 273K, all this becomes irrelevant to specify the pressure, entropy, etc, because all these variables are what we call state functions. State functions depend only on the state of the system.
Other quantities such as work (ww) and heat (qq), on the other hand, are not state functions. There is nothing like the work done or the heat in a system.in a process the amount of heat and work that is flowing in the system that connects specified initial and final states depend on how the process is carried out. Quantities that depend on the path followed between states are called path functions.
How is all this connected to differentials? Quantities whose values are independent of path are called state functions, and their differentials are exact (dPdP, dVdV, dGdG, dTdT...). Quantities that depend on the path followed between states are called path functions, and their differentials are inexact (dwdw, dqdq). when we integrate an exact differential, the result depends only on the final and initial points, but not on the path chosen. However, when we integrate an inexact differential, the path will have a huge influence in the result, even if we start and end at the same points.
Knowing that a differential is exact will help you derive equations and prove relationships when you study thermodynamics in your advanced physical chemistry courses. For example, you will learn that all the state functions we mentioned above are related through these equations:
dU=TdS−PdV
dH=TdS+VdP
dA=−SdT−PdV
dG=−SdT+VdP
Here, we introduced two new state functions: the enthalpy (HH), and the Helmholtz free energy (AA). they are state functions, just as the entropy and the free energy. explicitly, T, P, VT, P, V and SS are not constants. When discussed about gases, we learned that P, VP, V and TT are not independent functions. If two of these variables are changed the third, or in other words, we cannot independently vary the pressure, volume and temperature. The equation of state tells you how the three variables depend on each other.
For one mole of gas, the equation of state as a function can be written as
P=P (V, T) P=P (V, T), or as a function V=V (T, P) V=V (T, P), or as a function T=T (P, V) T=T (P, V).
In the same way, you cannot independently change the pressure, volume, temperature and entropy of a system. If you modify the pressure and temperature, the volume and entropy will change as well. To make this clear, we can re-write the equations above as:
dU=T (S, V) dS−P (S, V) dV
dH=T (S, P) dS+V (S, P) dP
dA=−S (T, V) dT−P (T, V) dV
dG=−S (T, P) dT+V (T, P) dP
H, AU, H, A and GG are all state functions, their differentials are exact. We can derive a few relationships just from this fact.
For example, we see that G=G (T, P) G=G (T, P), and because it’s total differential, by definition, is:
dG=(∂G∂T) PdT+(∂G∂P) TdP
we rapidly conclude that
(∂G∂T) P=−S(∂G∂T) P=−S
and
(∂G∂P) T=V(∂G∂P) T=V
With minimal math, we concluded that if we change the pressure of a system at constant temperature, the rate of change of the free energy equals the volume. At this point this does not mean so much, but can hopefully appreciate how knowing that GG is a state function is enough for you to derive a thermodynamic relationship!
−(∂S∂P) T=(∂V∂T) P
we derived this equation from the knowledge that GG is a state function. Why are these equations useful? Let’s see an example We can integrate this expression with TT constant to get:
∫P2P1dS=ΔS=−∫P2P1(∂V∂T) PdP (constant)
This equation tells you that the change in entropy in a system can be calculated by integrating (∂V∂T) P(∂V∂T) P data. This is extremely powerful, as we can easily measure temperature, pressure and volume in the lab, but we don’t have an instrument that directly measures entropy!
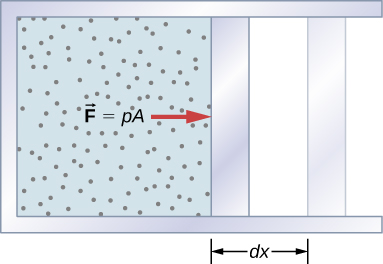
A force that is created from any source by work done from any source moving through an object through a displacement. How is thermodynamic system work? The figure shows a picture of a gas that is confined to a cylinder which has a movable piston at one end, if the gas expands against the piston, it exerts force through a distance and does work on the piston. When the piston compresses the gas inward as it moves inward, work is done, on the gas, the work related to such changes is as follows.
Let the gas pressure on the piston face be p. Then the force on the piston due to the gas is pA, where A is the area of the face. When the piston is pushed outward an infinitesimal distance dx, the magnitude of the work done by the gas
dW = F dx = pA dx
Since the change in volume of the gas is
dV = A dx
this becomes
dW = pdV
For a finite change in volume from V1 to V2
we can integrate this equation from V1 to V2 to find the net work:
W = 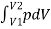
The work done by a confined gas in moving a piston a distance dx is given by
dW = Fdx = pdV
This integral is only meaningful for a quasi-static process, which means a process that takes place in infinitesimally small steps, keeping the system at thermal equilibrium.
This relationship can be plotted on a pV diagram of pressure versus volume, where the curve is the change of state. We can approximate such a process as one that occurs slowly, through a series of equilibrium states. The integral is interpreted graphically as the area under the pV curve (the shaded area of figure. Work done by the gas is positive for expansion and negative for compression.
When a gas expands slowly from V1 to V2 the work done by the system is represented by the shaded area under the pV curve.
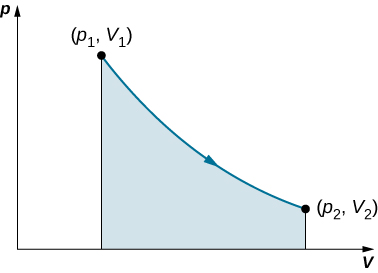
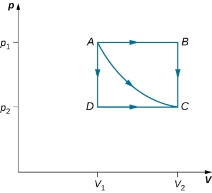
Consider the two processes involving an ideal gas that are represented by paths AC and ABC in figure.
The first process is an isothermal expansion, with the volume of the gas changing its volume from V1 to V2. This isothermal process is represented by the curve between points A and C. The gas is kept at a constant temperature T by keeping it in thermal equilibrium with a heat reservoir at that temperature. From figure and the ideal gas law,
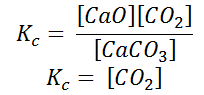
The paths ABC, AC, and ADC represent three different quasi-static transitions between the equilibrium states A and C.
The expansion is isothermal, so T remains constant over the entire process. Since n and R are also constant, the only variable in the integrand is V, so the work done by an ideal gas in an isothermal process is
Notice that if V2>V1 (expansion), W is positive, as expected.
The straight lines from A to B and then from B to C represent a different process. Here, a gas at a pressure  first expands isobarically (constant pressure) and quasi-statically from V1 to V2, after which it cools quasi-statically at the constant volume V2 until its pressure drops to
first expands isobarically (constant pressure) and quasi-statically from V1 to V2, after which it cools quasi-statically at the constant volume V2 until its pressure drops to  . From A to B, the pressure is constant at p, so the work over this part of the path is
. From A to B, the pressure is constant at p, so the work over this part of the path is
W = 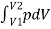 = p1
= p1 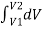 = p1 (V2-V1)
= p1 (V2-V1)
From B to C, there is no change in volume and therefore no work is done. The net work over the path ABC is then
W = p1(V2-V1) + 0 = p1 (V2-V1)
A comparison of the expressions for the work done by the gas in the two processes of (Figure) shows that they are quite different. This illustrates a very important property of thermodynamic work: It is path dependent. We cannot determine the work done by a system as it goes from one equilibrium state to another unless we know its thermodynamic path. Different values of the work are associated with different paths.
Internal Energy
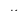

E int = 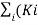 + Ui)
+ Ui)
where the summation is over all the molecules of the system, and the bars over K and U indicate average values. The kinetic energy Ki of an individual molecule includes contributions due to its rotation and vibration, as well as its translational energy miv2i /2 where vi
of an individual molecule includes contributions due to its rotation and vibration, as well as its translational energy miv2i /2 where vi is the molecule’s speed measured relative to the center of mass of the system. The potential energy Ui is associated only with the interactions between molecule i and the other molecules of the system. In fact, neither the system’s location nor its motion is of any consequence as far as the internal energy is concerned.
is the molecule’s speed measured relative to the center of mass of the system. The potential energy Ui is associated only with the interactions between molecule i and the other molecules of the system. In fact, neither the system’s location nor its motion is of any consequence as far as the internal energy is concerned.
In an ideal monatomic gas, each molecule is a single atom. Consequently, there is no rotational or vibrational kinetic energy and Ki = miv2i /2. Furthermore, there are no interatomic interactions (collisions notwithstanding), so Ui = constant, which we set to zero. The internal energy is therefore due to translational kinetic energy only and
E int = 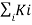 =
= 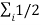 mi
mi 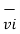 2
2
We know that the average kinetic energy of a molecule in an ideal monatomic gas is
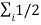 mi
mi 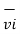 2 = 3/2 KB T
2 = 3/2 KB T
where T is the Kelvin temperature of the gas. Consequently, the average mechanical energy per molecule of an ideal monatomic gas is also 3kBT/2 that is,
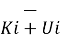 = Ki = 3/2 KBT
= Ki = 3/2 KBT
The internal energy is just the number of molecules multiplied by the average mechanical energy per molecule. Thus, for n moles of an ideal monatomic gas,
E int = n NA (3/2 KBT) = 3/2 n RT
The internal energy for a given quantity of an ideal monatomic gas is totally independent of pressure and volume and depends only on the temperature of the gas, in case of other systems the internal energy cannot be so easily expressed but however the internal energy can often be related to increase in temperature.
When two systems are kept in thermal contact, both eventually reach thermal equilibrium, the point at which both systems are at the same temperature as an example, suppose we mix two monatomic ideal gases. Now, the energy present per molecule of an ideal gas is proportional to its temperature. Therefore, when both the gases are mixed, energy is lost by the molecules that have hotter gas, and in case of colder gas energy is gained, this process continues until equilibrium is reached, at this point the average translational kinetic energy per molecule and the temperature is same for both the gases
The First Law of Thermodynamics
The Law of Conservation of Energy is also called as the First law of thermodynamics, according to the law which states that energy can neither be created nor destroyed, but can be changed or transferred from one form to the other. For example, turning on a light would seem to produce energy; however, it is electrical energy that is converted.
The expression of the first law of thermodynamics is, any change in the internal energy (∆E) of a system is given by the sum of the heat (q) that flows across its boundaries and the work (w) done on the system by the surroundings:
ΔE=q+wΔE=q+w
Two kinds of processes namely Heat and work are stated by the Law that can lead to a change in the internal energy of the system. Both heat and work can be quantified and measured, the change in the energy of a system must result in the surrounding change outside the system. In other words, energy cannot be created or destroyed. If heat flows into a system or the surroundings do work on it, the internal energy increases and the sign of q and w are positive. Conversely, heat flow out of the system or work done by the system (on the surroundings) will be at the expense of the internal energy, and q and w will therefore be negative.
Heat capacities
Thermodynamics in mainly concerned with heat, earlier heat was considered as a measure of an invisible fluid, caloric and present in any matter. The ability of a substance to hold this fluid was earlier referred to as the heat capacity of the system, the meaning however of today is that heat capacity is energy in transit. The development in thermodynamics and dependence of heat transfer on temperature changed the definition of heat.
Modern thermodynamics defines heat as the measure of the total internal energy of a system. In order to quantify the heat energy associated with matter and its dependence on temperature, two properties were defined. These properties were named as specific heat capacity and heat capacity of the system.
Heat Capacity Formula
Mathematically,
Q=CΔT
Where Q is the heat energy required to bring about a temperature change of ΔT and C is the heat capacity of the system under study.
Specific Heat Capacity
For thermodynamic studies the scientists wanted a quantity that did not depend on the quantity or size of matter, this led to Specific heat capacity. The Heat capacity is an intensive property, as it depends on the size and quantity of matter. Specific heat capacity for any substance or matter can be defined as the amount of heat energy required to raise the temperature of a unit mass of that substance by one degree Celsius. Mathematically it is given as:
Q=msΔT
Here Q is the amount of heat energy required to change the temperature of m (kg) of a substance by ΔT, s is the specific heat capacity of the system.
Thermodynamics continues to play a vital role in our lives directly or indirectly. Scientists and engineers use the laws of thermodynamics to design new processes for reactions that would have high efficiency and product yield. Chemical and mechanical engineers apply the concepts of thermodynamics for designing heat engines with high efficiency and better outputs.
As the body absorbs heat the temperature of the body rises, but when heat is withdrawn from the body it cools down, so the heat of the body decreases. The temperature of any body is the measure of its molecule’s kinetic energy. Heat capacity is the ratio of heat absorbed by a material to the temperature change. Therefore, the temperature change in a body is directly proportional to the heat transferred to the given body.
Definition of Molar heat capacity
The total amount of energy in the form of heat needed to increase the temperature of 1 mole of any substance by 1 unit is called molar heat capacity(C) of that substance. It also depends greatly on the nature, size and composition of a substance in a system.
q=n C ΔT
q is the heat supplied or needed to bring about a change in temperature(ΔT) in 1 mole of any given substance.
n is the amount of moles; the constant C is known as the molar heat capacity of the body of the given substance.
Cp: in a system, Cp is the amount of heat energy released or absorbed by a unit mass of the substance with the change in temperature at a constant pressure.in other words, under constant pressure it is the heat energy transfer between a system and the surroundings. So, Cp represents the molar heat capacity, C when pressure is constant. The change in temperature will always cause a change in the enthalpy of the system.
Enthalpy (ΔH) is the heat energy absorbed or released by the system, furthermore enthalpy change occurs during the change of phase or state of a substance.
For example, when a solid change to its liquid form (i.e., the change from ice to water) enthalpy change is called the heat of fusion. When a liquid change to its gaseous form (i.e., the change from water-to-water vapour) the enthalpy change is called the heat of vaporisation.
The system absorbs or releases heat without the change in pressure in that substance, then its specific heat at constant pressure, Cp can be written as:
Cp = [ dH/dT] p ----------------------------- 1
Where Cp represents the specific heat at constant pressure; dH is the change in enthalpy; dT is the change in temperature.
Cv: During a small change in the temperature of a substance, Cv is the amount of heat energy absorbed/released per unit mass of a substance, where volume does not change, in other words Cv is the heat energy transfer between a system and its surroundings, without any change in the volume of the system.
Cv represents the molar heat capacity C when the volume is constant. Under a constant volume, the volume of a substance does not change, so the change in volume is zero.
As the term is related to the internal energy of a system, which is total of both potential energy and kinetic energy of that system. The system absorbs or releases heat without change in volume of that substance, then its specific heat at constant volume Cv can be
CV = [ dU/dT] v --------------------------------------2
Where Cv represents the specific heat at constant volume, dU is the small change in the internal energy of the system; dT is the change in temperature of the systems.
Relationship between Cp and Cv
According to the first law of thermodynamics:
ΔQ= ΔU+ΔW where ΔQ is the amount of heat that is given to the system, ΔU is the internal energy and ΔW is the work done.
Therefore, we write
ΔQ=ΔU+PΔV as ΔW= PΔV……………………………3
Since ΔQ= nCpΔT and ΔU = nvCΔT
Therefore nCpΔT = nCpΔT + PΔV
We know that PV=nRT)
At T1 kelvin PV1=nRT1………………(a)
At T2 Kelvin PV2 = nRT2 -----------------(b)
Subtracting (a) from (b)
PV2 – PV1 = n RT2 – nRT1
P(V2-V1) = nR(T2-T1)
Where V2-V1 = ∆V
T2 – T1 = ∆T
Therefore P∆V = n R ∆T
Putting the value of P∆V we get
n Cp ∆T = n Cv∆T + n R ∆T
n Cp ∆T = n ∆T (Cv+ R)
Cp = Cv + R
Or Cp – Cv = R
The following relationship can be given considering the ideal gas behaviour of a Gas.
Cp-Cv = R
Where R is the universal gas constant.
Isothermal and adiabatic processes for ideal gas
The ideal gas law is the equation of state of a hypothetical gas, as illustrated in the figure, for an ideal gas there is no molecule-molecule interaction is present and only elastic collisions are allowed, although there are several limitations, it has a good approximation to the behaviour of various gases under different conditions. It was first stated by Émile Clapeyron in 1834 as a combination of Boyle’s law and Charles’ law.

Atoms and Molecules in a Gas, are typically and widely separated as shown in the diagram, because the forces between them are quite weak at these distances, they are often described by the ideal gas law.
Empirical Derivation
Boyle’s law states that pressure P and volume V of a given mass of confined gas are inversely proportional:
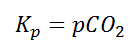
while Charles’ law states that volume of a gas is proportional to the absolute temperature T of the gas at constant pressure
V∝T.
By combining the two laws, we get

where C is a constant which is directly proportional to the amount of gas, n (representing the number of moles).
The proportionality factor is the universal gas constant, R, i.e., C = nR.
Hence the ideal gas law
PV=nRT.
Equivalently, it can be written as PV=NkT,
where k is Boltzmann’s constant and N is the number of molecules.
(Since N = nNA, and see that R=NAk, where NA is Avogadro’s number.)
The microscopic details are not considered by the empirical derivation However, the equation is therefore derived from the first principles of thermodynamics. (Which goes beyond the scope of this Atom).
Microscopic version
We have seen in the Atom on “Origin of Pressure” that
 where P is the pressure, N is the number of molecules, m is the mass of the molecule, v is the speed of molecules, and V is the volume of the gas. Therefore, we derive a microscopic version of the ideal gas law
where P is the pressure, N is the number of molecules, m is the mass of the molecule, v is the speed of molecules, and V is the volume of the gas. Therefore, we derive a microscopic version of the ideal gas law
Isotherms
An isothermal process is a change of a system in which the temperature remains constant: ΔT = 0.
An isothermal process is a change in the system in which the temperature remains constant: ΔT=0. This occurs typically when a system is in contact with an outside thermal system such as heat bath, and the change occurs slowly enough to allow the system to adjust continually to the temperature of the reservoir through heat exchange.
 In contrast, an adiabatic process occurs when a system exchanges no heat with its surroundings (Q=0), in ither words in an isothermal process, the value ΔT=0 but Q=0, while in an adiabatic process ΔT = 0 but Q = 0
In contrast, an adiabatic process occurs when a system exchanges no heat with its surroundings (Q=0), in ither words in an isothermal process, the value ΔT=0 but Q=0, while in an adiabatic process ΔT = 0 but Q = 0
For an ideal gas, the product PV (P: pressure, V: volume) is a constant if the gas is kept at isothermal conditions (Boyle’s law). According to the ideal gas law, the value of the constant is NkT, where N is the number of molecules of gas and k is Boltzmann’s constant.
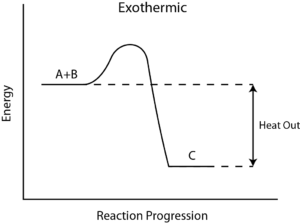
The family of curves generated by this equation is shown in the graph presented in. Each curve is called an isotherm. Such graphs are termed indicator diagrams first used by James Watt and others to monitor the efficiency of engines. The temperature corresponding to each curve in the figure increases from the lower left to the upper right.
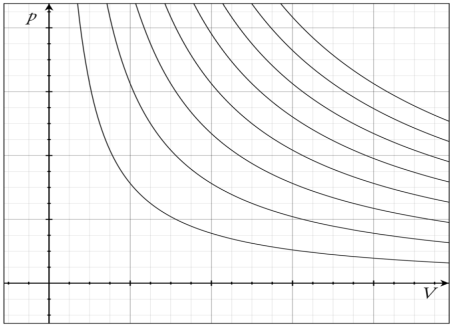
Isotherm of an ideal gas: several isotherms of an ideal gas on a PV diagram.
Calculation of Work
In thermodynamics, the work involved when a gas changes from state A to state B is simply:
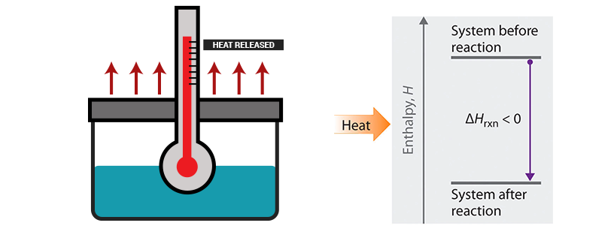
(This equation is derived in our Atom on “Constant Pressure” under kinetic theory. Note that P = F/A. This definition is consistent with our definition of work being force times distance.)
For an isothermal, reversible process, this integral equal the area under the relevant pressure-volume isotherm, and is indicated in blue in for an ideal gas. Again, P = nRT / V applies and with T being constant (as this is an isothermal process), we have:
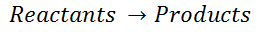
Work done by gas during expansion: The blue area represents ‘work’ done by the gas during expansion for this isothermal change.
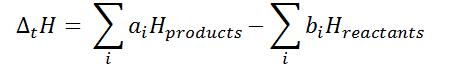
By convention, work is defined as the work the system does on its environment. If, for example, the system expands by a piston moving in the direction of force applied by the internal pressure of a gas, then the work is counted as positive. As this work is done by using internal energy of the system, the result is that the internal energy decreases. Conversely, if the environment does work on the system so that its internal energy increases, the work is counted as negative.
Difference Between Isothermal and Adiabatic Process
Following is the table explaining the isothermal vs adiabatic process:
Isothermal process | Adiabatic process |
An isothermal process is defined as one of the thermodynamic processes which occur at a constant temperature | An adiabatic process is defined as one of the thermodynamic processes which occur without any heat transfer between the system and the surrounding |
Work done is due to the change in the net heat content in the system | Work done is due to the change in its internal energy |
The temperature cannot be varied | The temperature can be varied |
There is a transfer of heat | There is no transfer of heat |
Relation between P-V, V-T and P-T for ideal gas
An essential condition is that
The system and surrounding must not be exchanging heat for which the walls should be insulated.
The process of expansion and compression so that the system should not be able to exchange heat with surroundings.
Relation between P and V
According to First law of thermodynamics
dQ = dU+ dW
For 1 mole of gas
dU= CvdT
dW = PdV
For adiabatic process dQ = 0
dQ =dU+dW
0=Cv dT+P dV
According to ideal Gas equation
PV= RT, differentiating on both sides we get
P dV+ V dP= R dT
dT = P dV+ V dP/R
Cv P dV+ V dP/R + P dV =0
Cv P dV+ Cv V dP + RP dV =0
Cv V dP + (Cv + R) P dV =0
Cv V dP + CP P dV = 0
Cv V dP/ Cv PV + CPP dV / Cv PV =0
dP/P + Cp dV/Cv V =0
dP/P + Cp / Cv dV/V =0
dP/P + 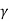 dV/V =0
dV/V =0
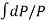 +
+ 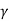
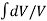 = C
= C
K is constant
This Equation gives the relationship between P and V of an ideal gas
Relation between P and T
We know that PV= RT for 1 mole of a gas
V = RT/P
Substituting the value of V

This equation gives the equation for P and T
Relation between V and T
We know that PV= RT for 1 mole of a gas
P = RT/V
Substituting the value of P in the equation

This equation gives the relation for V and T
Adiabatic processes for ideal gas
Adiabatically when an ideal gas is compressed, (Q=0) its temperature generally increases: when we consider an adiabatic expansion process, the ideal gas does not work and the temperature decreases, a few adiabatic compressions examples include compression occur in the cylinder of a car, where the gas-air mixture take place so quickly that there is no time for the mixture to exchange heat with its environment.
The figure below shows the adiabatic process in the free expansion of a gas, that is confined by a membrane to one side of a two-compartment that is thermally insulated container. When the membrane is punctured, the gases rushes to the other side of the container and therefore expand freely, Because the gas expands “against a vacuum” (p=0), it does no work, and because the vessel is thermally insulated, the expansion is adiabatic.
With Q=0 and W=0W=0 in the first law, ΔEint=0, ΔEint=0, so
Eint i = Eint f for free gas expansion.
The gas in the left chamber expands freely into the right chamber when the membrane is punctured.
If the gas is ideal, the internal energy depends only on the temperature. Therefore, when an ideal gas expands freely, its temperature does not change.
Adiabatic reversible expansion of ideal gas
In an Adiabatic process no heat enters or leaves the system, therefore, for a reversible adiabatic process the first law takes the form
dU = − PdV.
CV = (∂U/∂T) V. (when the, molar heat capacity is taken into account)
But the internal energy of an ideal gas depends only on the temperature and is independent of the volume (because there are no intermolecular forces), and so, for an ideal gas,
CV = dU/dT, and so we have dU = CVdT.
Thus, for a reversible adiabatic process and an ideal gas,
CVdT = −PdV. (The minus sign shows that as V increases, T decreases, as expected.) But for a mole of an ideal gas,
PV = RT = (CP − CV) T, or P = (CP − CV) T/V.
Therefore
CvdT=−(CP−CV) TdV/V
Separate the variables and write γ for CP/CV:
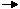
Integrate:
TVγ−1= constant.
This shows how temperature and volume of an ideal gas vary during a reversible adiabatic expansion or compression. If the gas expands, the temperature goes down. If the gas is compressed, it becomes hot. Of course, the pressure varies also, and the ideal gas conforms to the equation
PV/T = constant. On elimination of T, we obtain
PVγ= constant
On elimination of V, we obtain

Elementary idea of entropy and Clausius inequality.
Entropy
Solid state has the lowest or least entropy, the gaseous state has the highest entropy and the liquid state has the entropy that lies between the two.
Entropy is a state function. The changes in its value during any process, is called the entropy change.
ΔS = S2 -S1 = ∑S products – ∑S reactants
1) When a system absorbs heat, the molecules begin to move faster because the kinetic energy increases. therefore, disorder increases. Greater the heat absorbed, greater will be the disorder.
2) For equal amount of heat absorbed at low temperature, the disorder will be more than at high temperature. This proves that entropy change is inversely proportional to temperature.
ΔS = eve / T
Entropy change during a process is defined as the quantity of heat (q) absorbed isothermally and reversibly divided by the absolute Temperature (T) at which the heat is absorbed.
The entropy change between two specified states is the same whether the process is reversible or irreversible.
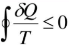
The inequity of the Clausius is a consequence of second law of thermodynamics
Q is he transfer of heat to and from the system
T is the absolute temperature at the boundary, the symbol 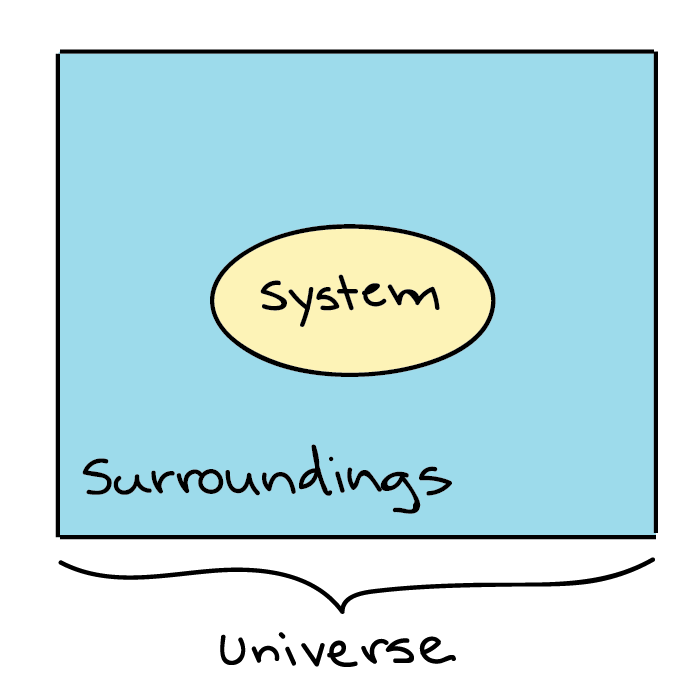 is the cyclic integral.
is the cyclic integral.
The Clausius inequality for a closed system states that the process is irreversible if TdS > dQ, and the process is reversible if TdS = dQ.
The entropy difference between any two states of a system they can be evaluated by the integral of its initial state and final state
Since entropy is a state function, the entropy change of the system for an irreversible path is the same as for a reversible path between the same two states.
For the reversible process the universe is brought back to its initial state without changing its properties. So, the entropy generation for the reversible process is zero. However, for the irreversible process, because of the disorder of the system some entropy is generated. the generated entropy is measure of the irreversibility. As a result of this the cyclic integral of the irreversible process is always less than zero.
The self-ionization of water deals with the process where the water ionizes to hydroxide ions and hydronium ions, this usually occurs on a limited scale. When two water molecules collide, there is a transfer of hydrogen ion from one molecule to the other. The products formed are hydronium ions that are positively charged and negatively charged hydroxide ions.
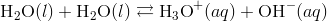
The simplified form of this equation is:
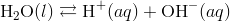
Kw is the ion-product of water symbol. The equilibrium constant for the self-ionization of water is referred to as the ion-product for water.
![K_w = [ text{H}^+ ][ text{OH}^- ]](https://glossaread-contain.s3.ap-south-1.amazonaws.com/epub/1642756539_249546.png)
The ion-product of water 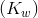 is mathematically the product of the concentration of hydroxide ions and hydrogen ions. Since water is in its pure form, it is not included in the ion-product expression the value of
is mathematically the product of the concentration of hydroxide ions and hydrogen ions. Since water is in its pure form, it is not included in the ion-product expression the value of 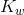 is very small, in accordance with a reaction that favours the reactants. At 25°C, the experimentally determined value of K w in pure water is 1.0 × 10 -14.
is very small, in accordance with a reaction that favours the reactants. At 25°C, the experimentally determined value of K w in pure water is 1.0 × 10 -14.
![K_w=[text{H}^+][text{OH}^-]=1.0 times 10^{-14}](https://glossaread-contain.s3.ap-south-1.amazonaws.com/epub/1642756539_411927.png)
In pure water, the concentrations of hydroxide ions and hydrogen ions are equal to one another, in case of pure water or any other aqueous solution, if this ratio exists it is said to be neutral. To find the molarity of each ion, the square root of 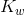 is taken.
is taken.
![[text{H}^+]=[text{OH}^-]=1.0 times 10^{-7} text{M}](https://glossaread-contain.s3.ap-south-1.amazonaws.com/epub/1642756539_505884.png)
An acidic solution, in this solution the concentration of the hydrogen ions is greater than that of the hydroxide ions. For example, hydrogen chloride ionizes to produce H + and Cl − ions upon dissolving in water.
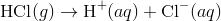
This leads to an increase in the concentration of H + ions in the solution. According to Le Châtelier’s principle, the equilibrium represented by 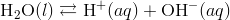 is forced to the left, towards the reactant. As a result, the concentration of the hydroxide ion decreases.
is forced to the left, towards the reactant. As a result, the concentration of the hydroxide ion decreases.
A basic solution, in this solution the concentration of hydroxide ions is greater than the concentration of hydrogen ions. Solid potassium hydroxide dissociates in water to yield potassium ions and hydroxide ions.
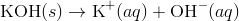
The increase in concentration of the OH − ions causes a decrease in the concentration of the H + ions and the ion-product of [H +] [OH −] remains constant
pH, pKa, pKb and pKh buffer solution
pH is a measure of the hydrogen concentration present in the solution, it measures how basic or acidic is an aqueous solution. The pH usually ranges from 0 to 14, the pH is said to be neutral when the value is 7, a pH value of less than 7 shows an acidic solution and a pH value above 7 represents a basic solution. The pH and organic content present in food form significant factors that signify not only the types of organisms that may survive during storage but also the activities of spoilage organisms.
Acid-base indicators are substances or dyes that change colour with pH, the pH scale shows the range of strength of acids and alkalis.
pH = - log [H+]
pOH = - log [OH-]
At 25 degrees Celsius:
pH + pOH = 14
Ka and pKa
Ka and Kb are related to each other through the ion constant for water, Kw:
Ka is dissociation constant for acid and pKa is simply the -log of this constant. Similarly, Kb is the dissociation constant for bases, while pKb is the -log of the constant. The acid and base dissociation constants are usually expressed in terms of moles per liter (mol/L). Acids and bases dissociate according to general equations:
In the formulas, A stands for acid and B for base.
A large Ka value indicates a strong acid because it means the acid is largely dissociated into its ions. A large Ka value also means the formation of products in the reaction is favoured. A small Ka value means little of the acid dissociates, so you have a weak acid. The Ka value for most weak acids ranges from 10-2 to 10-14.
The pKa gives the same information, just in a different way. The smaller the value of pKa, the stronger the acid. Weak acids have a pKa ranging from 2-14
Kb and pKb
Kb is the base dissociation constant. The base dissociation constant is a measure of how completely a base dissociates into its component ions in water.
A large Kb value indicates the high level of dissociation of a strong base. A lower pKb value indicates a stronger base.
pKa and pKb are related by the simple relation:
pKh buffer solution
A buffer solution is an aqueous solution consisting of a weak acid and its conjugated base mixture or vice versa. There is a minute change in its pH when a little or medium amount of strong base or acid is added to it and that is why it is used to avoid changes in the pH of a solution.
The pH value of the water solvent is 7, but if we add a few drops of HCl or NaOH solution, its pH decreases or increases respectively. Therefore, it is necessary to have solutions whose pH does not change even on the addition of strong alkalis or strong acids. Such solutions are called buffer solutions.
Buffer capacity is the capacity of a buffer solution to resist change in its pH. The equation is given by,
pH = pKa + log [Salt] / [Acid]
The pH of any acidic buffer solution is always less than 7 and the pH of any basic buffer solution is always greater than 7.
In many experiments, we need a buffer that maintains the solution at a specific pH. We may need an acidic or a basic buffer, depending on the experiment. The Henderson-Hasselbalch equation can help us choose a buffer that has the pH we want.
pH = pKa + log ([conj. base]/ [conj. acid]) With equal amounts of conjugate acid and base (preferred so buffers can resist base and acid equally), then
pH = pKa + log (1) = pKa + 0
Buffer Index, Buffer Capacity, Buffer Range
The buffer capacity on the other hand is expressed numerically and is equal to the minimum concentration of a strong base or a strong acid that brings a variation in the Buffer pH with one unit. The field of application of both notions is different: the buffer capacity is used in the quantitative chemical analysis and the buffer index in studying biological systems.
Buffers are generally characterised by pH range by which they maintain a more or less constant pH by their buffer capacity. the amount of strong acid or base that can be absorbed before the pH changes significantly. Though the pH range of a Buffer solution strongly depends on the chemical properties of the weak acid and weak base used to prepare the buffer (i.e., on KK), its buffer capacity depends solely on the concentrations of the species in the buffered solution. The more concentrated the buffer solution, the greater its buffer capacity.
The buffer solutions usually consist of a weak acid and its conjugate base. When H+ is added to a buffer, the protons (H+) are accepted by the acid’s conjugate base and absorbs the (H+) before the solution pH lowers significantly. Ina similar case, when OH- is added the weak acid donates a proton (H+) to its conjugate base, and thereby resists any increase in pH before shifting to a new equilibrium point. In biological systems, buffers prevent the fluctuation of pH via processes that produce acid or base by-products to maintain an optimal pH.
Each of the conjugate acid-base pairs have a specific pH range by which it works effectively as a buffer. The region of Buffering is about 1pH unit on both the sides of the pKa of the conjugate acid. The midpoint of the buffering region occurs when one half of the acid reacts to dissociation and the proton donor concentration(acid) is equal to the proton acceptor (base).
In other words, the pH of the equimolar solution of acid (e.g., when the ratio of the concentration of acid and conjugate base is 1:1) is equal to the pKa. This represents the point in the titration that is halfway to the equivalence point. This region is the most effective for resisting large changes in pH when either acid or base is added.
pH of a Buffer Solution
In chemistry, pH is a measure of the hydrogen ion (H+) concentration in a solution. The pH of a buffer can be calculated from the concentrations of the various components of the reaction. The balanced equation for a buffer is:
HA⇌H++A−HA⇌H++A−
The strength of a weak acid is usually represented as an equilibrium constant. The acid-dissociation equilibrium constant (Ka), which measures the propensity of an acid to dissociate, for the reaction is:
Ka=[H+] [A−][HA]Ka=[H+] [A−][HA]
The greater [H+] x [A–] is than [HA], the greater the value of Ka, the more the formation of H+ is favoured, and the lower the pH of the solution.
Idea of role of buffer solutions in day-to-day life
Maintenance of life
Most of the processes that include biochemical reactions, rely on a relatively small pH range. The body also uses buffer solutions for a constant pH maintenance.
For example, blood contains a carbonate/bicarbonate buffer that keeps the pH close to 7.4.
Biochemical Assays
Most of the enzyme assay or any enzyme activity depends on pH, therefore the pH during an enzyme process must be constant.
In shampoos.
Many of the shampoos either use citric acid/sodium citrate to shampoo to sustain a slightly acidic "pH balance".
This provides a balance with the detergents present in the shampoo.
In baby lotions.
The Ph in baby lotion s around 6, this helps in prevention of bacteria within diapers and also prevents diaper rash.
In the brewing Industry
Before fermentation begins, buffer solutions are added, this helps to prevent the solution from becoming acidic and also spoil the product.
In the textile Industry.
Many dyeing processes use buffers to maintain the correct pH for various dyes.
In laundry detergents.
Many laundry detergents use buffers to prevent their natural ingredients from breaking down
Dissociation constant of acids and bases
For a generalized acid dissociation
HA (aq) + H2O ⇌ A-(aq) + H3O+(aq)
The equilibrium constant will be
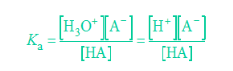
Where Ka is the acid dissociation constant. In dilute solutions it is assumed that the concentration of liquid water remains essentially constant when an acid is dissolved.
A weak base, represented by B, reacts with water to form an equilibrium solution of ions.
 B(aq) + H2O HB+ (aq) +OH- (aq)
B(aq) + H2O HB+ (aq) +OH- (aq)
The Equilibrium expression for this general reaction is
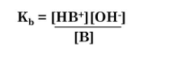
The higher the value of Kb the stronger the base.
Solubility product and its applications in salt analysis
The solubility product is known by the symbol Ksp, this solubility constant is the equilibrium constant for the dissociation of a solid substance into an aqueous solution.
The solubility product is an equilibrium constant and its value is dependent on temperature, due to increased solubility the Ksp increases with an increase in temperature.
Solubility is defined the property of a solute to completely dissolve in a solvent to form a solution. The solubility of the ionic compounds (which disassociate to form cations and anions) in water varies to a great deal. Some of the compounds show high solubility and are even able to absorb moisture from the atmosphere, but few other are highly insoluble.
Solubility depends on a number of parameters amongst which lattice enthalpy of salt and solvation enthalpy of ions in the solution are of most importance
Salts are classified on the basis of their solubility in the following table.
Category I | Soluble | Solubility > 0.1M |
Category II | Slightly soluble | 0.01M< Solubility<0.1M |
Category III | Sparingly soluble | Solubility < 0.1M |
Common Effect
The statement of the common ion effect can be written as follows –in a solution, where there are several species interacting with each other through a chemical equilibrium process, an increase in the concentration of one of the ions that is dissociated in the solution by the addition of another species that contain the same ions leads to an increase in the degree of association of ions.
An example of the common ion effect can be observed when gaseous hydrogen chloride is passed through a sodium chloride solution, leading to the precipitation of the NaCl due to the excess of chloride ions in the solution (brought on by the dissociation of HCl).
This effect cannot be observed in the compounds of transition metals. This is because the d-block elements have a tendency to form complex ions. This can be observed in the compound cuprous chloride, which is insoluble in water. This compound can be dissolved in water by the addition of chloride ions leading to the formation of the CuCl2– complex ion, which is soluble in water.
HSAB concept
For thousands of years people have known that vinegar, lemon juice, and many other foods taste sour. eventually, it was not until a few hundred years back that it was discovered why these foods taste sour – because they are all acids. The term acid, in fact, comes from the Latin term acere, which means "sour". While there may be different definitions of acids and bases, In the seventeenth century, An Irish writer and amateur chemist Robert Boyle first labelled substances as either acids or bases (he called bases alkalies), according to the following characteristics:
Acids: Acids become less acidic when mixed with bases they taste sour, are corrosive to metals, change litmus (a dye extracted from lichens) to red.
Bases: feel slippery, change litmus blue, and become less basic when mixed with acids.
While Boyle and others failed to explain why acids and bases behave the way they do, the first reliable definition of acids and bases was not be proposed until 200 years later.
In the late 1800s, the Swedish scientist Svante Arrhenius suggested that water can dissolve many compounds by separating them into their individual ions. Arrhenius suggested that acids are compounds that contain hydrogen and can dissolve in water to release hydrogen ions into a solution. For example, hydrochloric acid (HCl) dissolves in water as follows:
HCl H2O→ H+(aq) + Cl-(aq)
Arrhenius defined bases as those substances that can dissolve in water to release hydroxide ions (OH-) into the solution. For example, a typical base, sodium hydroxide (NaOH) was suggested by Arrhenius:
NaOH H2O→ Na+(aq) + OH-(aq)
The Arrhenius definition of acids and bases explains many things about Acids and Bases. Arrhenius’s theory corelates how all acids have similar properties to each other (and, conversely, why all bases are similar): since all acids release H+ into solution (and all bases release OH-). The Arrhenius definition clearly explains Boyle’s observation that bases and acids counteract each other. This idea, that a base can make an acid weaker, and that an acid can make a base weaker, is called neutralisation.
Neutralization
As you can see from the equations, acids release H+ into solution and bases release OH-. If we were to mix an acid and base together, the H+ ion would combine with the OH- ion to make the molecule H2O, or plain water:
H+(aq) + OH-(aq) → H2O
The neutralisation reaction of an acid with a base will always form water and a salt, as shown below:
Acid Base Water Salt
HCl + NaOH → H2O + NaCl
HBr + KOH → H2O + KBr
Though Arrhenius helped explain the fundamentals of acid/base chemistry, unfortunately his theories have limits. For example, the Arrhenius definition does not explain why some substances, such as common baking soda (NaHCO3), this compound act like a base even though they do not contain hydroxide ions.
In 1923, the Danish scientist Johannes Bronsted and the Englishman Thomas Lowry published an independent but similar paper that was defined in Arrhenius’ theory. According to Bronsted’s, "... acids and bases are substances that are capable of releasing or taking up hydrogen ions, respectively." The Bronsted-Lowry definition has a widespread concept of the Arrhenius of acids and bases.
The Bronsted-Lowry definition of acids and bases is very similar to the Arrhenius definition: Any substance that can release a hydrogen ion is an acid. (According to the Bronsted definition, acids are seldom mentioned to as proton donors because an H+ ion, hydrogen minus its electron, is simply a proton).
The Brønsted definition of bases is, however, very different from the Arrhenius definition. The Bronsted theory of base is defined as any substance that can concede a hydrogen ion. In other words, a base is the opposite of an acid. NaOH and KOH, as seen above, would still be considered bases because they can accept an H+ from an acid to form water. However, the Bronsted-Lowry definition also explains the reasons why substances that do not contain OH- can still act like bases. Baking soda (NaHCO3), for example, acts like a base by accepting a hydrogen ion from an acid as illustrated below:
Acid Base Salt
HCl + NaHCO3 → H2CO3 + NaCl
In this example, the carbonic acid formed (H2CO3) undergoes rapid decomposition to water and gaseous carbon dioxide, and so the solution bubbles as CO2 gas are released.
Key takeaway
In a chemical equilibrium, the forward and reverse reactions occur at equal rates, and the concentrations of products and reactants remain constant, in broad terms, thermodynamics deals with the transfer of energy from one place to another and from one form to another. The key concept is that heat is a form of energy corresponding to a definite amount of mechanical work. The equilibrium established between the unionised molecules and the ions in the solution of weak electrolytes is called ionic equilibrium.
References:




