Unit - 3
Chemistry of Aliphatic hydrocarbons
Formation of alkenes and alkynes by elimination reaction
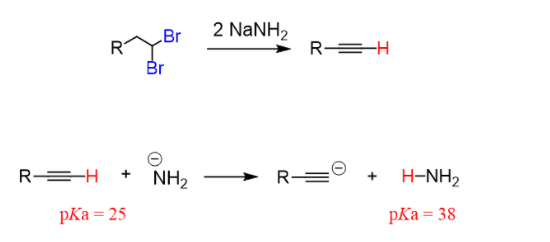
Alkynes are prepared by elimination of alkyl dihalides:
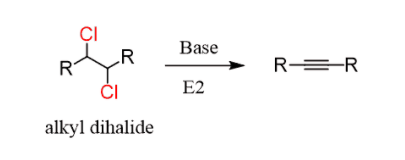
It is similar to the preparation of alkenes and the mechanism is by the E2, the only difference being, two halogens are needed as the triple bond id formed by two consecutive elimination reactions:
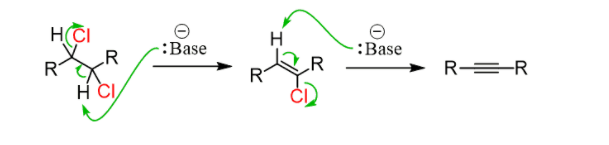
Hydroxides and alkoxides form the regular bases for the first elimination. However, a stronger base is needed to achieve an elimination of the vinyl halide. Most often Sodium amide (NaNH2), dissolved in liquid ammonia (NH3), is used:
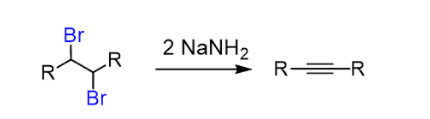
The two halogens can be on the same (geminal dihalide) carbon or on neighbouring (vicinal dihalide) carbons.
To distinguish these, you can go with geminal as being “jammed” on one carbon:
More scientifically, it comes from the Latin geminus – “twin.”
Depending on the structure of the dihaloalkane, the elimination reaction results in either an internal or terminal alkyne:
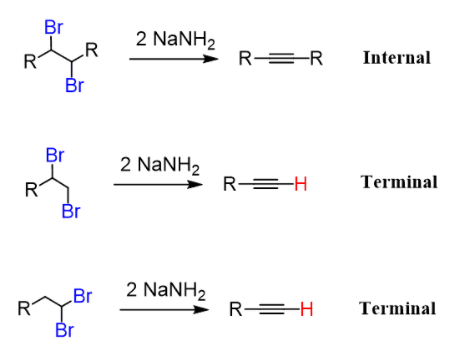
The hydrogen atom on the triple bonded carbon is an added advantage for the preparation of terminal alkynes.
The equilibrium shifts to the right, as it is driven by the deprotonation of the sodium amide, the hydrogen here has a pKa of 25.
Totally three equivalents of sodium amide are needed: two equivalents for the two E2 eliminations and one to deprotonate the terminal alkyne.
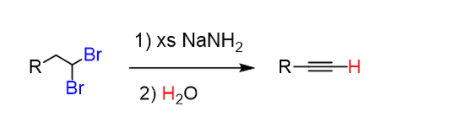
Once the reaction is complete, the alkynide ion is protonated back so as to regenerate the terminal alkyne. This is the work-up of the reaction and is achieved with water since the acidity of water allows to perform this acid-base reaction:
Even when an internal alkyne is prepared, an excess of sodium amide is often used to make sure the reaction goes to completion. Therefore, the water work-up is used in any case the protonate the alkynide ion and also quench the possible excess of NaNH2.
Overall, the reaction is shown like this:
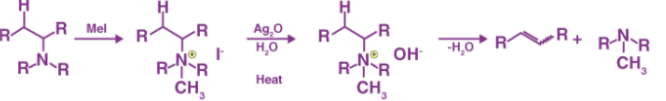
Hofmann’s elimination is the process of creating tertiary amines and alkenes from the treatment of quaternary ammonium with excess methyl iodide, and the treatment of the resulting compound with silver oxide, water, and heat. The product formed from the treatment of quaternary ammonium with excess methyl iodide is called quaternary ammonium iodide salt. The replacement of the iodine by a hydroxyl anion is followed by an elimination reaction to form the alkene as well.
What is Hofmann Elimination?
The process of producing tertiary amines and alkenes from quaternary ammonium is called Hofmann elimination. This process can also be referred to as exhaustive methylation. The Hofmann rule states that the major alkene product is the least substituted and least stable product when it comes to asymmetrical amines. The Hofmann elimination process is named after its discoverer, the German chemist August Wilhelm Von Hofmann. The Hofmann elimination can be illustrated as follows.
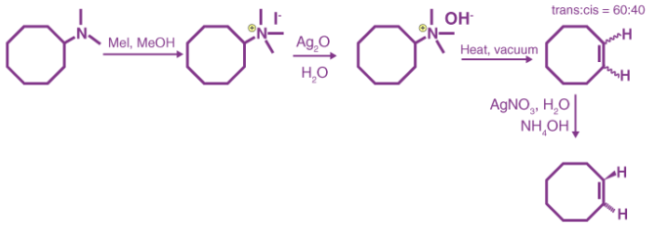
It can be noted that methyl iodide is used in excess because it has no beta hydrogens and therefore cannot compete in the elimination reaction. If the given alkyl group contains two different sets of beta hydrogens which are available for the elimination process, the alkene isomer having less substituted double bond is formed as the major product. An important example of the Hofmann elimination process or the exhaustive methylation process is the synthesis of trans-cyclooctene which is shown below.
It is important to note that the neutral NR3 group acts as a better leaving group than the anionic NH2– or the NR2– groups. The outcome of the reaction is also affected by steric effects caused by large leaving groups.
Hofmann Elimination Mechanism
The Hofmann elimination mechanism or the exhaustive methylation mechanism begins with the attack of the amine with a beta-hydrogen on the methyl iodide to form the ammonium iodide salt. Now, the iodide reacts with the silver oxide to form silver iodide. This silver iodide is insoluble and hence is precipitated out of the solution. A silver oxide ion deprotonates the water to form a hydroxide ion. This mixture is now heated. The heat facilitates the elimination reaction, the hydroxide now picks up the beta-hydrogen from the ammonium ion. It also releases an amine to give the final alkene product. The mechanism can be illustrated via the following steps:
Step 1
The ammonium iodide salt is formed from the treatment of the amine with a beta-hydrogen or quaternary ammonium with excess methyl iodide, as shown below. 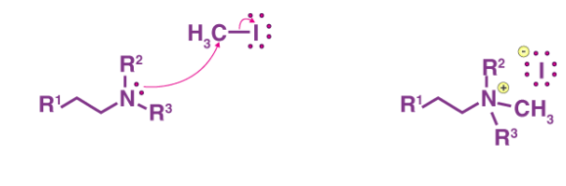
Step 2
The substitution of the iodide ion with the hydroxide ion is achieved by the reaction between the iodide and silver oxide and the successive deprotonation of water by the silver oxide ion. This step can be illustrated as follows.
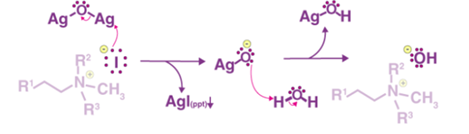
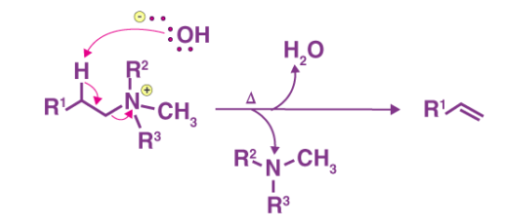
Step 3
The mixture is heated to facilitate and elimination reaction and form the required product.
Thus, the required olefin product is achieved. The tertiary amine product is also formed. The Hofmann elimination process can be categorized as an elimination reaction as well as an olefination reaction
Elimination reaction is a type of reaction is mainly used to transform saturated compounds (organic compounds which contain single carbon-carbon bonds) to unsaturated compounds (compounds which feature double or triple carbon-carbon bonds).
Besides, it is an important method for the preparation of alkenes.
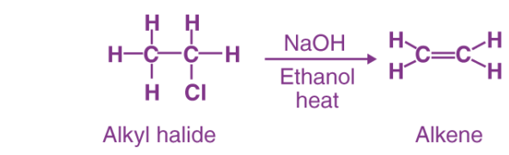
An elimination reaction is a type of chemical reaction where several atoms either in pairs or groups are removed from a molecule. The removal usually takes place due to the action of acids and bases or action of metals. It can also happen through the process of heating at high temperatures.
E1 Reaction
- In the E1 mechanism which is also known as unimolecular elimination, there are usually two steps involved – ionization and deprotonation.
- During ionization, there is a formation of carbocation as an intermediate. In deprotonation, a proton is lost by the carbocation.
- This happens in the presence of a base which further leads to the formation of a pi-bond in the molecule.
- In E1, the reaction rate is also proportional to the concentration of the substance to be transformed.
- It exhibits first-order kinetics.
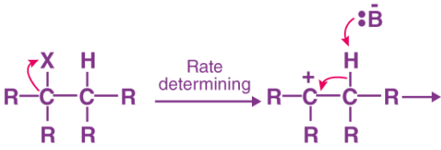
E 1 mechanism shares the features of the SN1 reaction. The initial step is the formation of a carbocation intermediate through the loss of the leaving group. This slow step becomes the rate-determining step for the whole reaction.
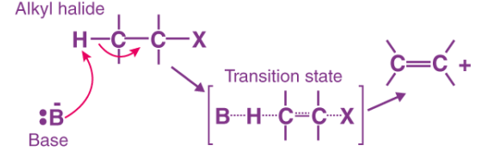
E2 Reaction
- In an E2 mechanism which refers to bimolecular elimination is basically a one-step mechanism.
- Here, the carbon-hydrogen and carbon-halogen bonds mostly break off to form a new double bond.
- However, in the E2 mechanism, a base is part of the rate-determining step and it has a huge influence on the mechanism.
- The reaction rate is mostly proportional to the concentrations of both the eliminating agent and the substrate.
- It exhibits second-order kinetics.
The E2 mechanism can generally be represented as below. In the below-mentioned representation, B stands for base and X stands for the halogen.
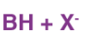
The rate of the E2 reaction is
Rate = k[RX][Base]
So, the reaction rate depends on both the substrate (RX) and the base involved. In the elimination reaction, the major product formed is the most stable alkene.
E1cb reaction
The third possibility is that the beta carbon is deprotonated first (form B–H, break C–H). In a limited case it results in an anionic intermediate, from the substrate, which is a “conjugate base”.
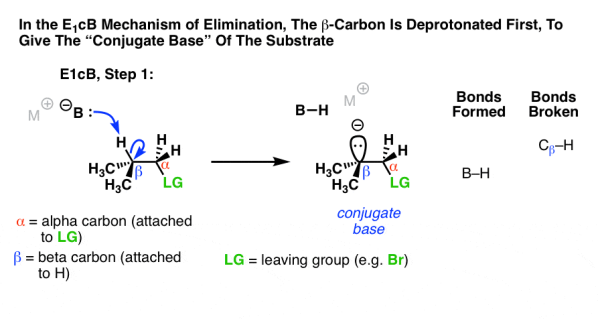
In the first step an anion is produced, by the deprotonation process, this reaction mainly occurs when the C–H bond is relatively acidic, when electron withdrawing groups like a ketone, nitrile (CN), or nitro group (NO2) and when the alpha carbon is adjacent to an electron, however these groups stabilise the negative charges. These groups stabilize negative charge.
In the second step, the pair of electrons from the conjugate base then displaces the leaving group, forming a pi bond [form C–C pi, break C–LG].
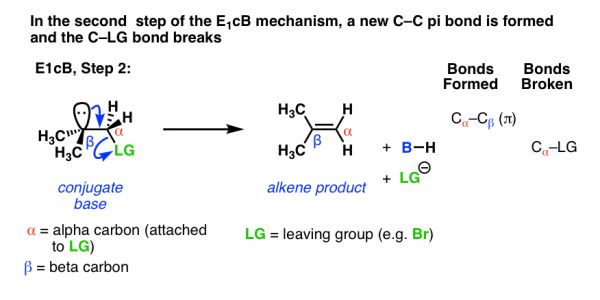
In the (many) cases of the E1cB where the second step is the slow step, you can imagine that this step would be made slower by a relatively poor leaving group like (HO-), (CH3O-) or even fluorine
The key difference between Saytzeff and Hofmann rule is that Saytzeff rule indicates that the most substituted product is the most stable product, whereas Hofmann rule indicates that the least substituted product is the most stable product.
In organic elimination reactions, the prediction of the end product depends on the Saytzeff rule and Hofmann rule therefore they are very important, for a particular organic chemical reaction, these rules can predict the nature of the final product, on the basis of substitution of the final product, e.g., most substituted or least substituted product
What is Saytzeff Rule?
Saytzeff rule, is a very important aspect as it determines the final product as the most substituted product, it is mainly an empirical rule, it is also named as Zaitsev’s rule. In the elimination reaction, the final product is determined by this rule and this rule helps in predicting the substitution of the final alkene product. According to the carbon atom where the double bond forms in the final alkene product, we can define Saytzeff rule as “the alkene that forms via elimination of hydrogen from the beta-carbon having the least hydrogen substituents”. Therefore, the most substituted final product is the major product of the chemical reaction, which also turns out to be the most stable product.
For example, 2-iodobutane contains an iodide group at the second carbon of the carbon chain. When 2-iodobutane is treated with strong acids like potassium hydroxide, the product formed is 2-butene as the major product and the minor product is 1-butene. Here, the carbon atom that has the fewest hydrogen substituents is the third carbon atom; therefore, the elimination of a hydrogen atom occurs from this carbon which gives 2-butene.the most hydrogen substituents are present on the first carbon atom, therefore elimination of hydrogen from the first carbon atom forms the minor product 1-butene. The reaction is as follows:
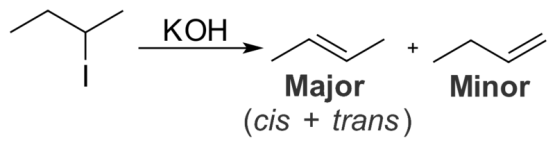
Figure: An Elimination Reaction that Occurs According to Saytzeff Rule
What is Hofmann Rule?
Hofmann rule is an empirical rule this rule predicts, the final product as the least substituted product of a particular reaction. That means; a chemical reaction takes place according to this rule if the majority of the final product is the least substituted olefin (alkene product). According to the carbon atom where the double bond forms in the final alkene product, we can define Hofmann rule as “the alkene that forms via elimination of hydrogen from the alpha-carbon having the most hydrogen substituents”.
Let’s look at an example:
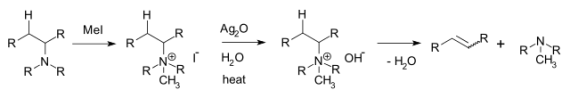
Figure: A Reaction that Occurs According to Hofmann Rule
During the creation of quaternary ammonium and alkene via the reaction between a tertiary amine and excess methyl iodide, the least substituted product of alkene forms as the major product.
These are group of alkenes that belong to the unsaturated hydrocarbons, which means that one molecule of alkene at least contains one double bond. Addition reactions are seen in alkenes due to the presence of pi bonds, in the addition reactions an electrophile attacks the carbon-carbon double bond resulting at the formation of double bonds. Reaction occurs when HBr is added to unsymmetrical alkenes in the presence of peroxide, 1-bromopropane is formed contrary to 2-bromopropane (according to Markovnikov’s rule). This reaction is better known as anti-Markovnikov addition or Kharash effect after the name of M. S. Kharash who first observed it. This reaction is also known as Kharash effect or peroxide effect.
Anti Markovnikov addition reaction which represents the alkenes, but is an exception to the Markovnikov’s rule. It is one of the few reactions following free radical mechanism in organic chemistry in place of electrophilic addition as suggested by Markovnikov. This reaction is observed only with HBr, not with HCl or HI.
Mechanism of Anti Markovnikov addition
Anti Markovnikov addition reaction is found to follow a free radical mechanism. The peroxide compound involved helps in the generation of free radicals. A general mechanism of anti-Markovnikov addition reaction is discussed below:
- By the homolytic cleavage of the peroxide compound free radical is generated.
- Through hemolysis, the free radical that is generated on the hydrogen halide, to form hydrogen halide.
- Formation of alkyl radical, by hemolysis, by the attack of the halide radical that is generated, on the alkyl molecule.
- Attack of a generated alkyl radical on hydrogen halide to form alkyl halide through homolytic cleavage of hydrogen halide bond.
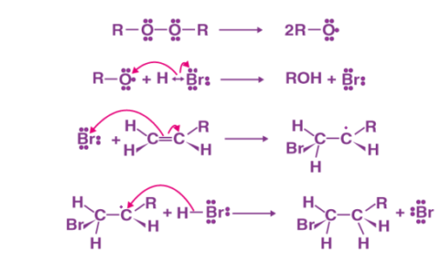
Fig: Anti Markovnikov addition mechanism
Demercuration
In the first step, the mercuric acetate, Hg(OAc)2, dissociates and forms into a mercuric cation:
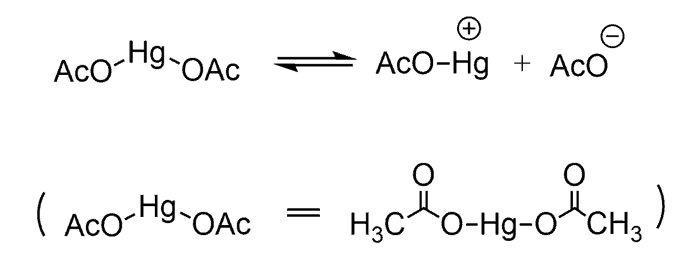
The cation that is formed is more electrophilic and gets attacked by the double bond., resulting in the formation of carbocation that is substituted. The key difference comes when comparing the structurers obtained in each reaction:
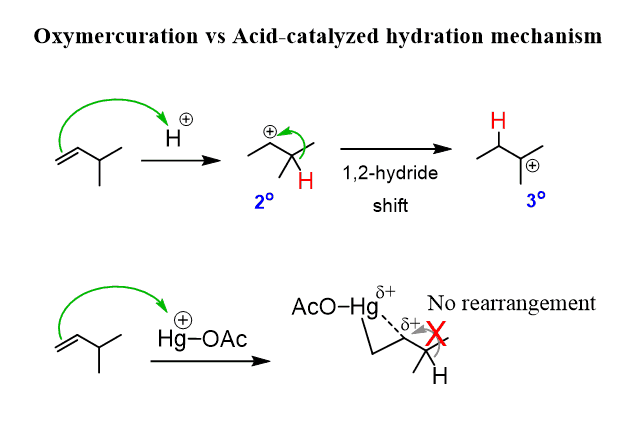
In the first reaction, the intermediate formed is anormal carbocation that can undergo rearrangements. However, when the π bond attacks a mercuric cation, the resulting intermediate is resonance-stabilized three-membered ring named as a mercurinium ion.
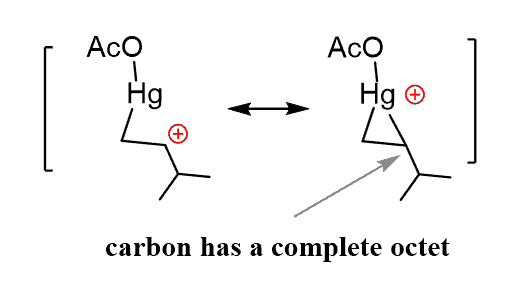
The carbon structures have a complete octet unlike the carbocations, and the two resonance structures, of which one can be stable. In the resonance-hybrid, the carbon and the mercury have partial positive charges.
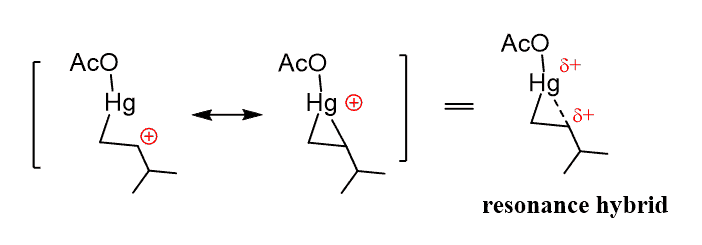
Unlike the carbocation, this carbon now bears a partial positive charge (δ+), rather than a full positive charge and does not readily undergo a rearrangement reaction.
However, as the partial positive charge is present and also the presence of the ring strain, the reaction is still possible, and gets attacked by water. The attack occurs at the more substituted carbon of the bridged mercurinium ion since it better stabilizes the partial positive charge:
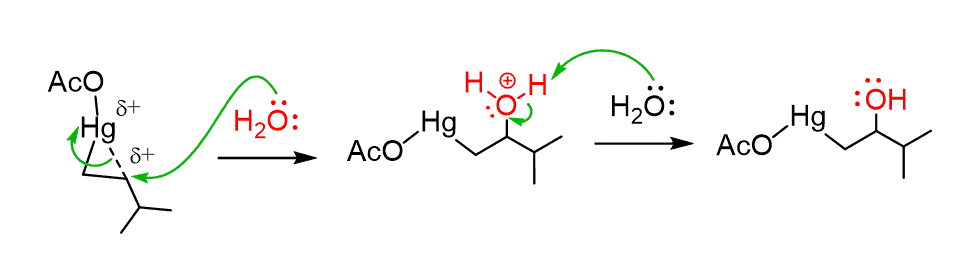
The major product formed is alcohol as the Markonikov addition leads to the more substituted alcohol, as the attack takes place on the more substituted position.
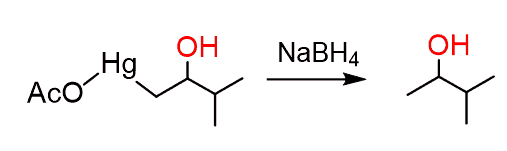
The mercury is then removed using sodium borohydride (NaBH4) which reduces the acetoxy mercury group and replaces it with hydrogen. This is described as demercuration:
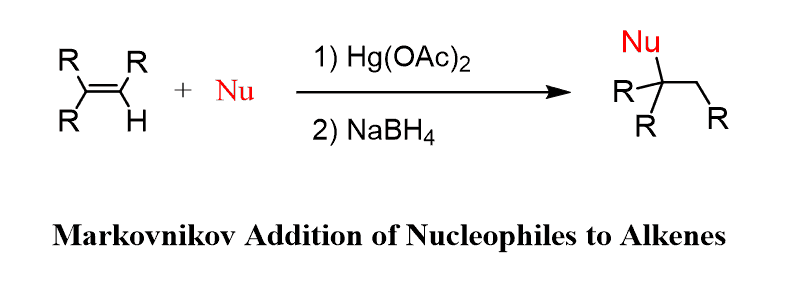
The electrophile that can be used as a reactant for nucleophilic reactants such as
Alcohols, thiols, and amines, is the mercurinium ion So, we can put together the following equation as a general guide for the Oxymercuration-Demercuration:
The Stereochemistry of Oxymercuration-Demercuration
There is no stereochemical control in this reaction because the addition of the mercuric ion and the hydrogen in the demercuration step occurs both as syn and anti-orientation. As a result, the new stereogenic center forms in R and S configuration leading to a racemic mixture:
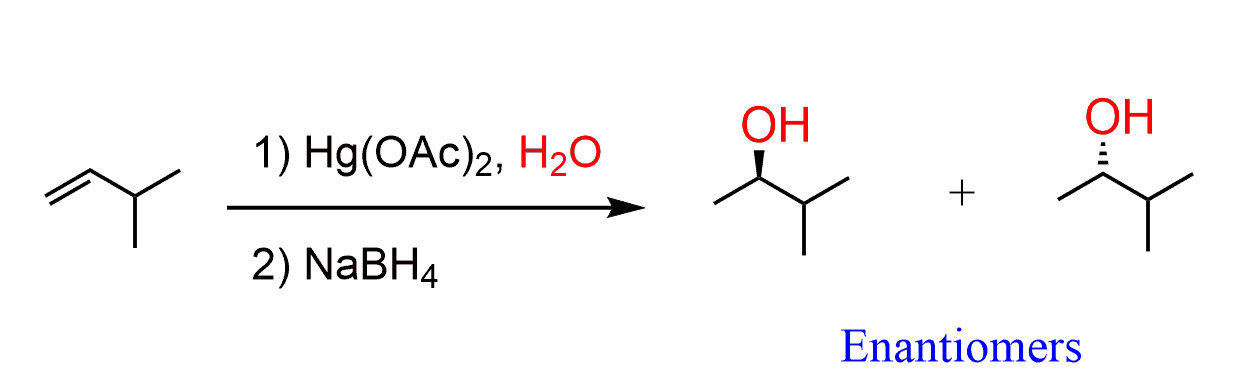
Hydroboration-Oxidation
Hydroboration-oxidation converts alkenes into alcohols:
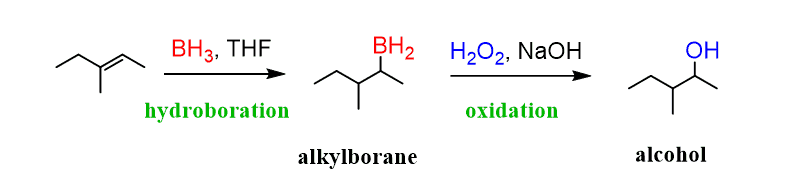
THF (tetrahydrofuran) is the solvent that is used to stabilize the dimer of BH3 that’s flammable, toxic, and explosive gas:
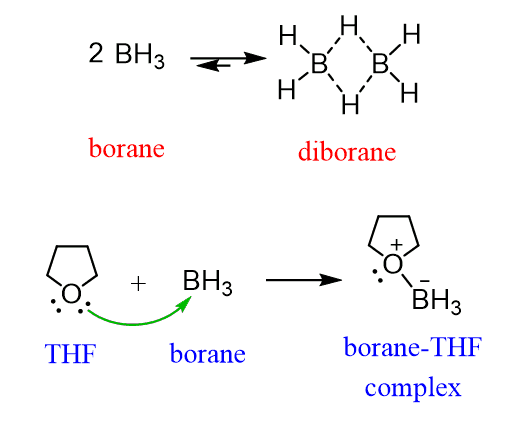
It is a some-steps transformation that begins from the addition of borane (BH3) to the alkene. This is called hydroboration and it is an electrophilic addition to the alkene. Boron is a Lewis acid and accepts the electrons of the π bond:
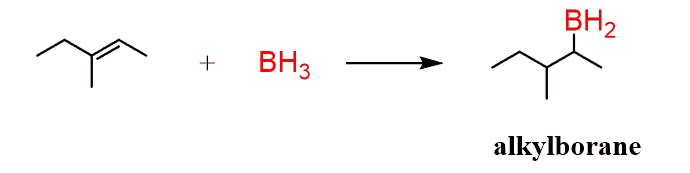
This is a concerted mechanism, which means that all the bonds in a reaction are broken and formed at the same time.
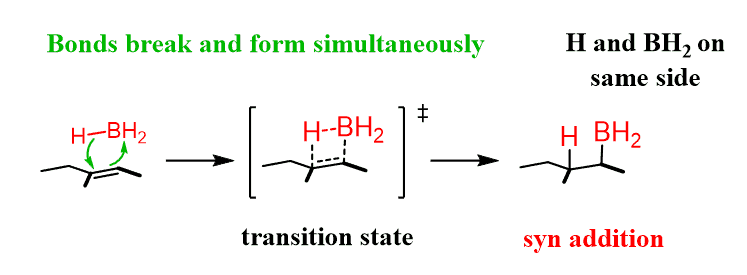
The resulting compound formed has still two hydrogens and the reactions undergoes repetition several times resulting in the conversion to trialkyl borane. Although, steric factors may prevent this especially for bulky alkyl groups.
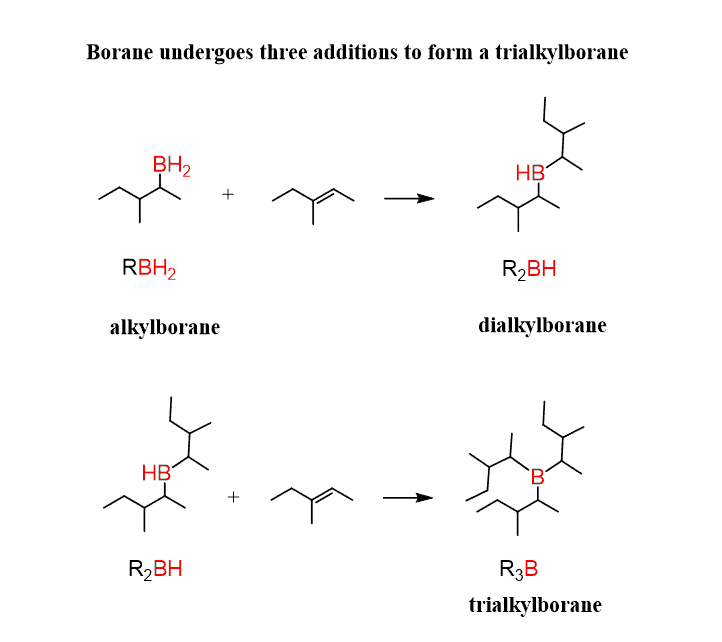
The trialkyl borane formed in the second part of the reaction, gets oxidised to alcohol.
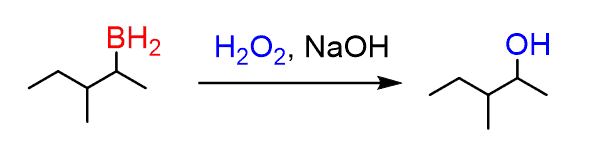
This formation of alcohol takes a few steps, and begins with the deprotonation of the hydrogen peroxide:
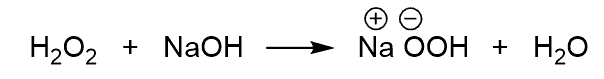
The resulting hydrogen peroxide ion does a nucleophilic attack at the boron:
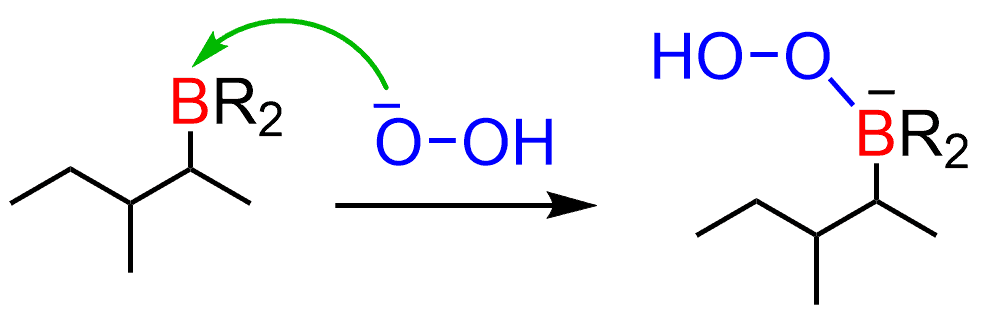
After this, a 1,2-alkyl shift occurs reducing the electron density on the boron:
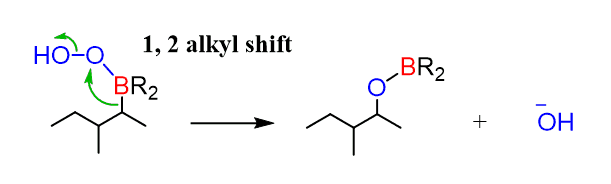
Another nucleophilic attack, this time by –OH, followed by loss of the alkoxide ion and its protonation produces the target alcohol:
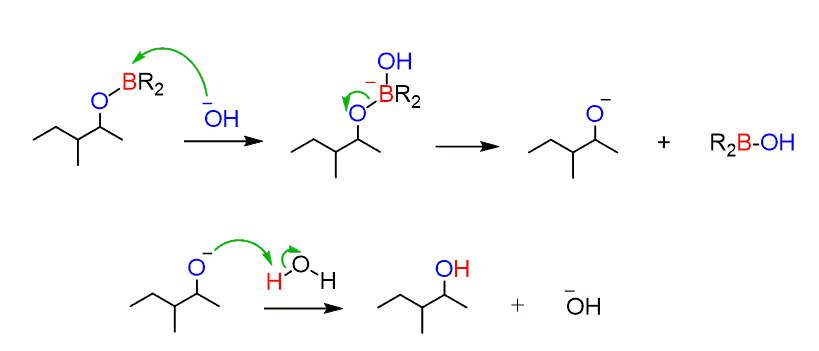
In summary, hydroboration-oxidation places an OH group on the less substituted carbon of the alkene:
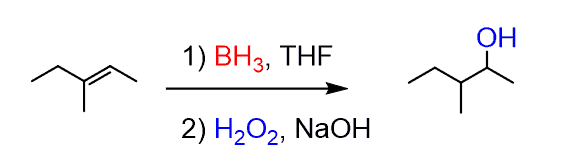
In few cases when BH3 is used the addition reaction occurs first and as only one H is attached to the boron, it is needed for the entire conversion., the oxidation of alkyl boranes is often shown with the monoalkyl borane instead of the trialkyl borane. Commercially available dialkyl boranes are used. The most common one is 9-Borabicyclo[3.3.1]nonane or 9-BBN:
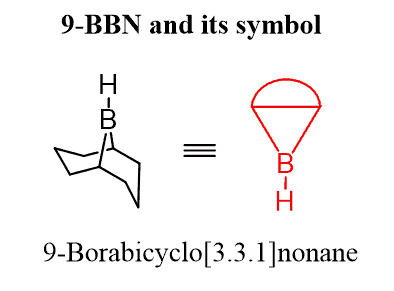
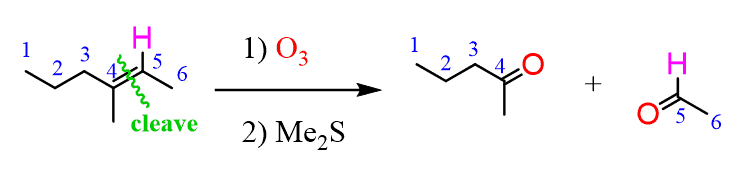
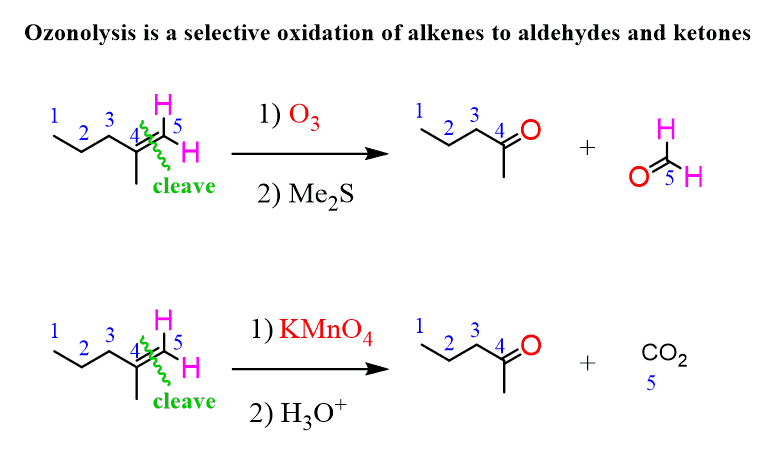
Ozonolysis is an efficient way of oxidizing alkenes ( and alkynes as well) into their corresponding aldehydes and ketones:
The advantage of the ozonolysis is when compared to other cleavage reactions they do not over-oxidize the alkene to carboxylic acid unlike, for example, the potassium permanganate (KMnO4):
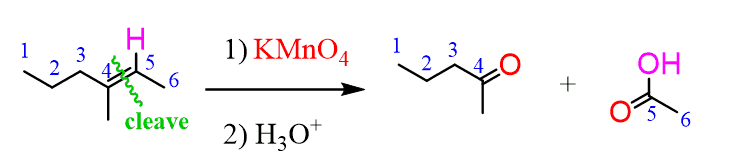
Even for terminal alkenes, it is possible to obtain the formaldehyde instead of carbon dioxide which is formed when strong oxidizing agents are used:
A similar principle is applied to cyclic compounds, when the product in ozonolysis has to be determined counting the number of carbons and replacing them with carbonyls
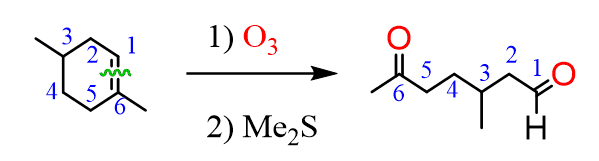
Ozonolysis Mechanism
The process of oxidation is obtained by bubbling ozone into the reaction mixture at a temperature which is very low (-78 oC). The first step is the formation of a cyclic intermediate called molozonide (initial ozonide) which quickly rearranges into the more stable ozonide:
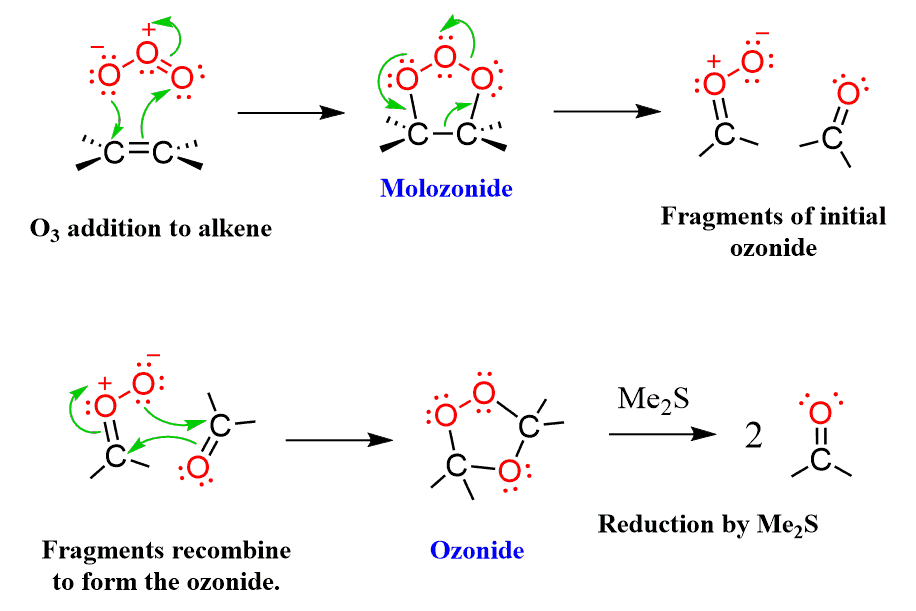
The last step involves a reducing agent dimethyl sulfoxide (Zn and Acetic acid are also used) is used. Sulfur, being a polarizable atom, induces a partial positive charge on one of the connected oxygens and attacks it cleaving the weak O-O bond:
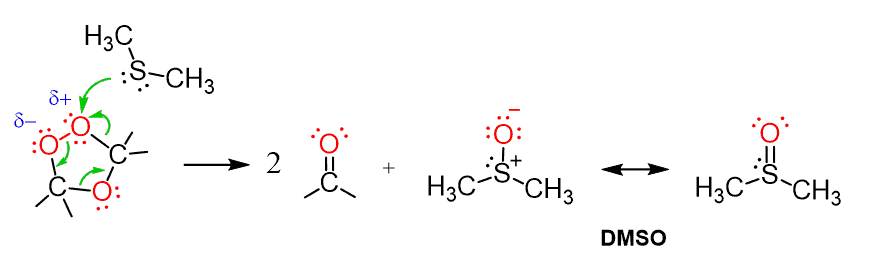
As a result, the carbonyls are formed together with DMSO
Reduction
Hydrogenation is one of the most important addition reactions in alkenes, it is a reaction where the alkene undergoes reduction to form alkane. Two hydrogen atoms are added across the double bond of an alkene in the hydrogenation reaction, this process leads to saturated alkane. The process of hydrogenation in double bonds are thermodynamically favourable as it forms a more stable lower energy product, in more defined terms the energy of the product is less than the energy of the reactant. Thus, it is exothermic (heat is released). The heat released is called the heat of hydrogenation, which is an indicator of a molecule’s stability.
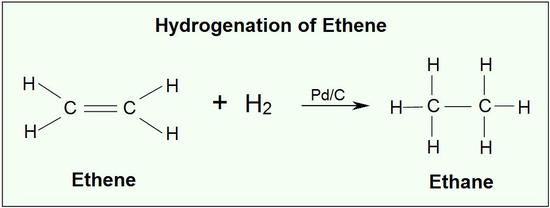
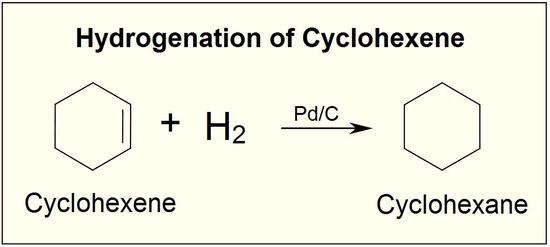
Although the hydrogenation of an alkene is a thermodynamically favourable reaction, it will not proceed without the addition of a catalyst.
A catalyst lowers the activation energy needed for the reacting molecules to reach the transition state. The addition of a catalyst enables the hydrogenation reaction to occur, that otherwise would not.
Insoluble metals form the common catalyst in this process, they include Pd-C, platinum in the form PtO2, and nickel in the form Ra-Ni. The H-H bond in H2 cleaves, in the presence of a catalyst and each hydrogen clings to the metal catalyst leading to the formation of metal-hydrogen bonds., the metals catalyst absorbs the alkene on its surface. A new C-H bond is formed, as the hydrogen atom is transferred to the alkene, a second hydrogen atom is transferred forming another C-H bond, at this position two hydrogen atoms are added to carbons across a double bond.
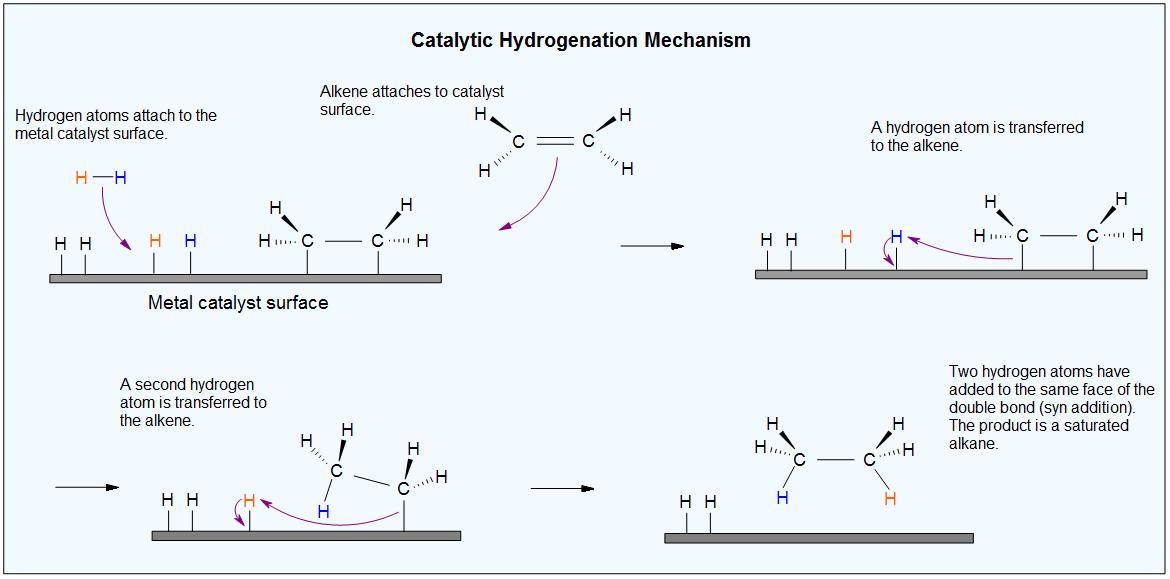
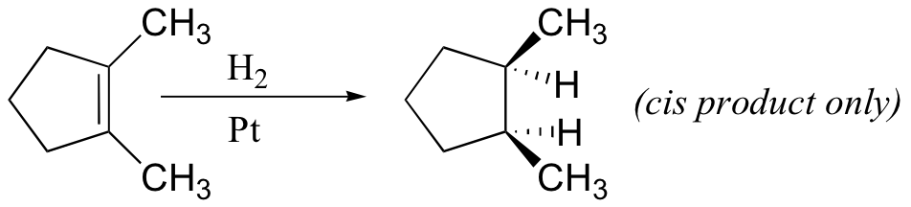
Because this reaction takes place on a planar surface, addition of hydrogen occurs on the same face of the double bond – a syn addi
Addition, in other words. The catalytic hydrogenation of 1,2-dimethylcyclopentane will yield, for example, the cis dimethylcycloalkane product, with little or no formation of a trans product.
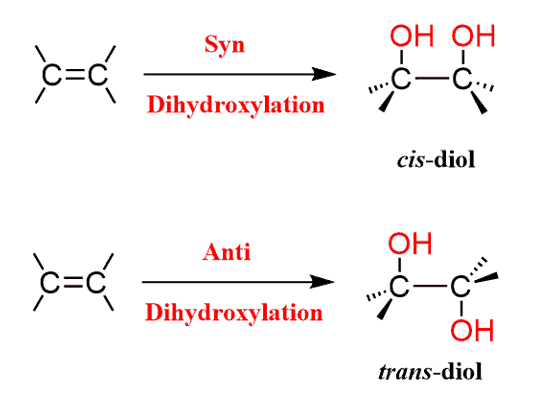
Dihydroxylation is the addition of two OH groups to an Alkene:
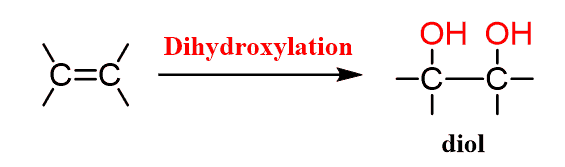
This can be achieved in several ways depending on the structure of the target diol, mainly, whether we need it in a cis or trans configuration:
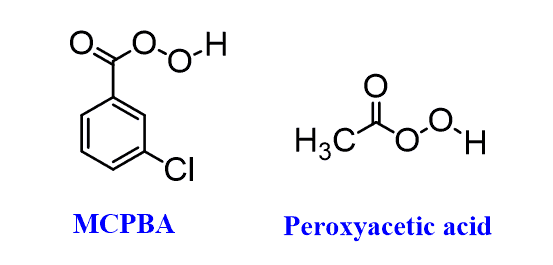
The first focus would be on the alkene being converted into an epoxide, this is achieved by peroxyacids like MCPBA (meta-chloroperoxy benzoic acid) or Peroxyacetic acid:
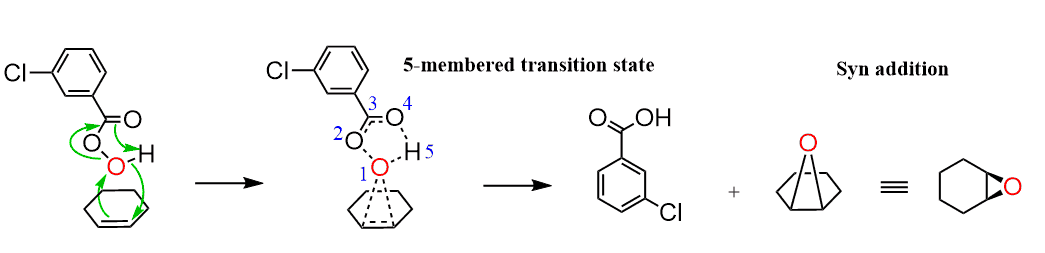
The mechanism of epoxidation is similar to the formation of alonium and oxymercuration, discussed above
The electron deficient oxygen present in peroxide is attacked by the p electrons of the π bond. In addition, the O-O bond is relatively weak which also makes the bond-cleavage easier:
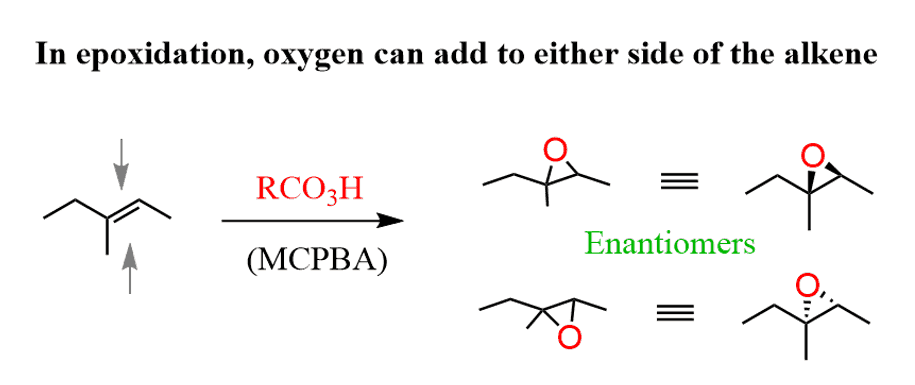
The epoxidation is a syn addition the oxygen is added on either side of the alkene, resulting in the formation of enantiomers when substituted alkenes react with acid peroxides:
Anti-Dihydroxylation Mechanism
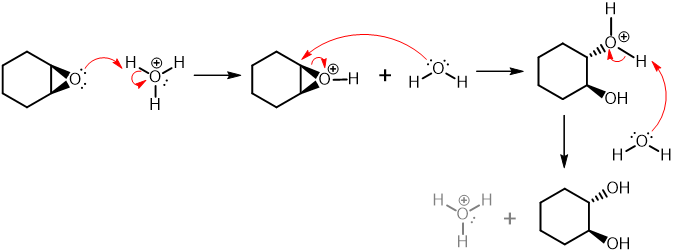
The first step in anti-dihydroxylation is epoxidation of the alkene using a peroxyacid (a.k.a. Peracid--MCPBA is the most common peroxyacid and is often specifically used). In the reaction the second step, is ring opening of the epoxy intermediate, this reaction is acid catalysed, with water as the nucleophile. The epoxide formed is protonated followed by attack with water, this back side attack is the ultimate reason that this reaction results in anti addition.
Step 1 - Epoxidation
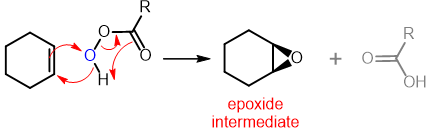
Step 2 - Acid-Catalysed Ring Opening of an Epoxide
The conjugated diene namely 1,3 butadiene, the compound the reactions of 1,3-butadiene are reasonably typical of conjugated dienes. The compound usually undergoes reactions with alkene, these may include catalytic hydrogenation or radical and polar additions, but it does so more readily than most alkenes or dienes that have isolated double bonds. Furthermore, the products frequently are those of 1,2 and 1,4 addition:
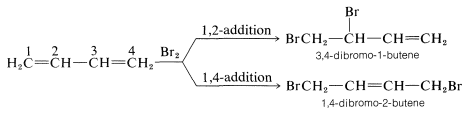
It’s not only in halogens that the addition products of Formation of both 1,2- and 1,4-addition occurs, but this process also occurs in electrophiles like hydrogen halides. The reaction of 1,3-butadiene with hydrogen chloride is shown below (13-1). The first step is similar to the alkenes that involves the formation of carbocation. However, with 1,3-butadiene, if the proton is added to C1C1 (but not C2C2), the resulting cation has a substantial delocalization energy, with the charge distributed over two carbons due to type IV resonance. Attack of Cl⊖Cl⊖ as a nucleophile at one or the other of the positive carbons yields the 1,2- or the 1,4- addition product:
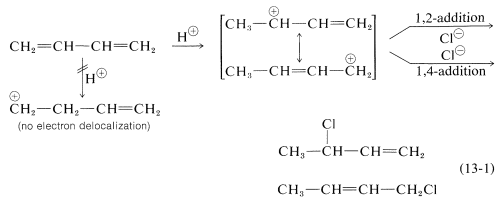
The interesting point to note is that the reactions of 1,2 and 1,4 additions compete with one another and the ratio of products depends upon factors like the temperature, the solvent, and also on the total time of reaction. Since formation of carbocation is reversible, a lot depends on the reaction time of the reaction., and the ratio of products at equilibrium need not be the same as the ratio of the rates of attack of Cl⊖Cl⊖ at C1C1 and C3C3 of the carbocation. This is another example of a difference in product ratios resulting from kinetic control vs equilibrium control
.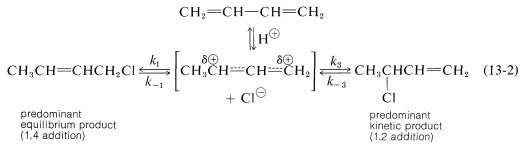
The fact is that at low temperatures the 1,2 product predominates because it is formed more rapidly, and the back reactions, corresponding to k−1k−1 or k−3k−3, are slow (Equation above). However, at equilibrium11 the 1,4 product is favoured because it is more stable, not because it is formed more rapidly.
The Diels-Alder reaction is an organic chemical reaction that is very important as it includes a substituted alkene and a conjugated diene, the substituted alkene is called as dienophile, the reaction results in a substituted derivative of cyclohexene. The Diels-Alder reaction is a very good example of pericyclic reactions which proceed via concerted mechanisms (i.e., all bond breakage and bond formation occurs in a single step).
In the year 1928, German chemist Otto Diels and Kurt Alder discovered the reaction, they were also rewarded the Nobel Prize in Chemistry. The Diels-Alder reaction can be used to form six-membered rings since there is a simultaneous construction of two new carbon-carbon bonds.
An illustration of the reaction is provided below
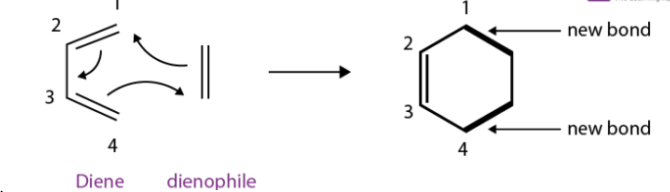
From the above reaction, it is noticed that the two pi bonds are converted into two sigma bonds, due to the concerted bonding of two independent pi-electron systems. The Diels-Alder reaction involves the shift of four pi electrons of the diene and two pi electrons of the dienophile.
This reaction is used in the production of vitamin B6. The reverse reaction (also called the retro-Diels-Alder reaction) is used in the production of cyclopentadiene on an industrial scale.
Mechanism of Diels-Alder Reaction
The reaction is thermodynamically favourable as the pi bonds are converted into strong sigma bonds; the reaction is favoured by electrophilic dienophiles with electron-withdrawing groups attached to them. It is also favoured by nucleophilic dienes with electron-donating groups in them. Given below are a few examples for good dienes and dienophiles for the Diels-Alder reaction
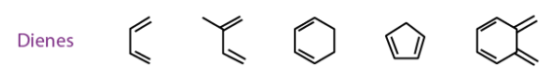
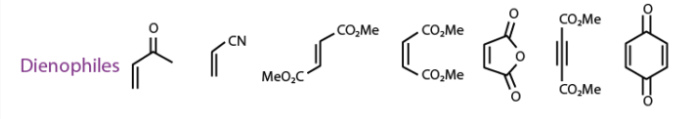
The reaction moves forward in a single step cycloaddition reaction as the Since the Diels-Alder reaction mechanism is concerted, here, two unsaturated molecules combine to form a cyclic adduct. There is a net reduction in bond multiplicity. All the bond formations and bond breakages happen simultaneously. Given below is an illustration of the simple reaction mechanism.
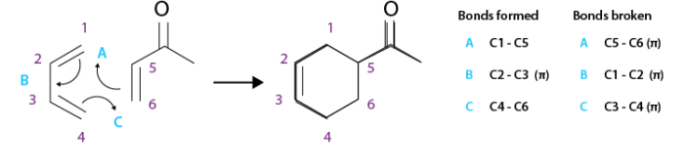
Alkynes are one of the simplest hydrocarbons. They have general formula of CnH2n-2. Alkynes belong to the family of unsaturated hydrocarbons that is; they contain both sigma and pi bond linkages between carbon and hydrogen

Alkyne molecule consist of at least one triple bond present between two carbon atoms, for example ethyne (CH≡CH). It reacts with bases that are strong like sodium metal and sodamide (NaNH2) resulting in the formation of sodium acetylide along with the liberation of dihydrogen gas. The acidity of alkenes is shown in the reaction, as the alkynes liberate dihydrogen gas on reaction with bases.
HC ≡ CH + Na → HC ≡ C– Na+ + 1/2H2
Relative Acidity of Alkynes
The acidity of alkynes is determined by their ability to loose a hydrogen atom to form alkynideions. Thus, alkynes act as Brønsted-Lowry acids. In alkynes the triple bond in the carbon atoms is “sp” hybridized. Due to the high percentage of “s” character (50%) in alkynes, the “sp” hybridized orbitals of carbon atom in alkynes exhibit high electronegativity. The electronegativity attracts the C-H bond of alkynes to a large range. Thus, alkyne molecules can easily lose hydrogen atom forming alkynide ions. Hence, we can say that the hydrogen atom attached to the triply bonded carbon atom is acidic in nature
The acidity of alkynes is greater than the acidity of alkanes and alkenes as the carbon atom in alkanes and alkenes are “sp3” and “sp2” hybridized respectively. Hence, these molecules contain a smaller percentage of “s” character in comparison to alkynes. Thus, the electronegativity of the carbon atom in these cases is lesser than alkynes. Hence, alkanes and alkenes don’t show the reactions with bases to liberate hydrogen gas. It can further be noted that only hydrogen atom attached to a triply bonded carbon atom are acidic not the other hydrogen atoms in the alkyne chain. The general trend in acidity is seen as:
HC≡CH > H2C=CH2> CH3–CH3
HC≡CH>CH3–C≡CH>>CH3–C≡C–CH3
Alkenes exhibit wide range of electrophilic addition reactions. Addition of hydrogen halides such as hydrogen bromide and hydrogen chloride are an example of electrophilic addition reactions of alkenes. The general trend of hydrogen halide is given as: HI >HBr> HCl. For symmetrical alkenes such as ethene it is quite easier to predict the end product in comparison to unsymmetrical alkenes such as propene. For example:
CH2 = CH2 + H−Br → CH3−CH2−Br
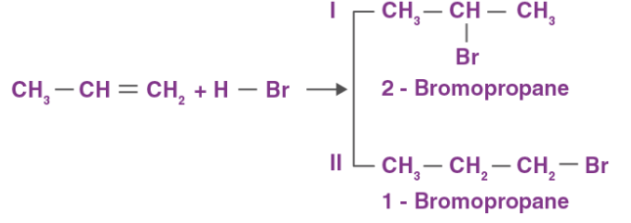
Markovnikov rule states that negative part of the adding molecule gets attached to that carbon atom which possesses lesser number of hydrogen atoms. Thus, 2-bromopropane is the expected product by this rule. This can further be explained with the help of mechanism of electrophilic substitution reactions of alkenes. The general mechanism is explained below:
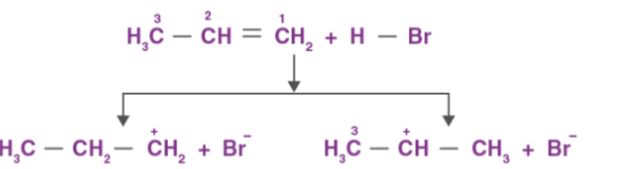
An electrophile, H+ is generated from hydrogen bromide which attacks on double bond to form carbocation.
Since, the secondary carbocation is more stable than the primary carbocation, the secondary carbocation predominates the formation of ions.
Finally, Br– attacks the carbocation to form alkyl halides

Nucleophilic addition reaction of alkynes is generally catalysed by the presence of heavy metal salts such as those of mercury and barium, the function of mercury ions (Hg2+ ) or barium ions (Ba2+) is to form 7r-complex by withdrawing the electron density from the ᴨ cloud
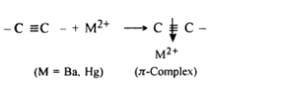
As a result the electron density in the triple bond gets considerably reduced and therefore, the nucleophilic attack is facilitated
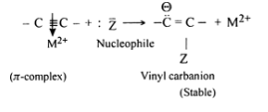
In other words, the pi complex formation is responsible for the nucleophilic addition reaction of alkynes. For example, alkynes undergo following nucleophilic addition reactions:
- The addition of water undergoes following nucleophilic addition reactions
- The addition of HCN in the presence of Ba (CN)2
- The addition of acetic acid in the presence of Hg2+ ion
Hydration of alkynes is another method for the preparation of carbonyl compounds. This method involves the addition of water to alkynes in the presence of mercuric sulphate and sulphuric acid to form aldehydes or ketones as the final product.
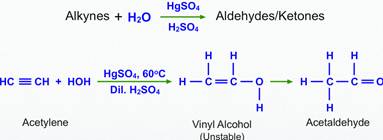
A terminal alkyne is defined as an alkyne having a hydrogen substituent (Fig. 1). This hydrogen substituent is acidic and can be removed with strong base (e.g. Sodium amide NaNH2) to produce an alkynide (Fig. 2). This is an example of an acid–base reaction.
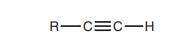
Terminal alkyne

Reaction of a terminal alkyne with a strong base
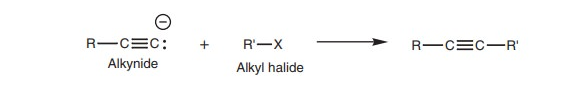
Reaction of an alkynide ion with an alkyl halide
Alkylation
Once the alkynide has been formed, it can be treated with an alkyl halide to produce more complex alkynes (Fig. 3). This reaction is known as an alkylation as far as the alkynide is concerned, and is an example of nucleophilic substitution as far as the alkyl halide is concerned.
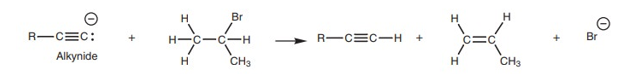
Elimination of HBr
This reaction works best with primary alkyl halides. When secondary or tertiary alkyl halides are used, the alkynide reacts like a base and this results in elimination of hydrogen halide from the alkyl halide to produce an alkene.
The class of hydrocarbons having a ring structure are the Cycloalkanes, the ring is formed as a result of their saturated nature. Additionally, they have three compounds of alkane that are present in the structure that help in the formation of the ring. They have the general formula CnH2n , n represents the number of carbon atoms present in the organic compound.
These consist of carbon-hydrogen bonds and also carbon to carbon single bonds where these carbon atoms join forming a ring or in the shape a cyclic structure. The smallest cycloalkane is Cyclopropane and most of the members belonging to this class are stable in nature.
Some common examples of cycloalkanes are the cyclopentane, Cyclobutane, cyclohexane, and cycloheptane, cyclooctane, etc as shown below in the image. The number of carbon atoms present in the compound decides the structure of cycloalkane. For example, Cyclobutane is a saturated hydrocarbon with four carbon atoms and the hydrocarbon with three atoms in its structure is called cyclopropane.
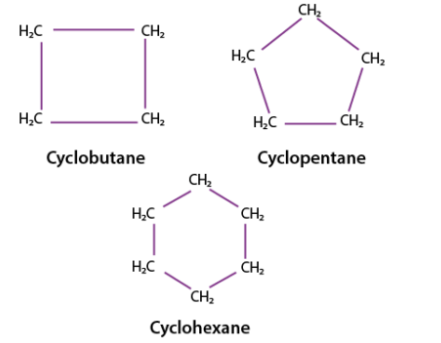
All the cycloalkanes do not have the same relative stability, but among all of them the most stable one is cyclohexane and is also experimentally proven whereas cyclobutane and cyclopropane is less stable This difference in relative stability is due to ring strain, which comprises angle strain and torsional strain.
Angle strain is the result of deviation from ideal bond angles caused by inherent structural constraints (such as ring size).
Torsional strain is the result of repulsive dispersion forces that cannot be relieved due to restricted conformational mobility.
Adolf von Baeyer, in1885 a German scientist had proposed the Baeyer’s strain theory to explain the relative stability of cycloalkanes.
Postulates of Baeyer’s strain theory:
- All the cycloalkanes are planar, and the atoms of carbon that constitute the ring lie in the same plane.
- Angle strain is the strain in the ring, that is caused in the deviation of bond angle from the normal tetrahedral angle (109o 28′).
- The instability increases with increase in the angle strain,
- Higher the stability of the ring, greater would be its ease of formation.
The angle strain in various cycloalkanes was calculated by Baeyer, the three atoms in cyclopropane occupy the corners of an equilateral triangle therefore, the C-C-C bond angle is 60o. The normal tetrahedral angle is 109o 28′.
In the formation of cyclopropane, the normal angle is 109o 28′ has been reduced to 60o. Hence, the angle strain in cyclopropane is calculated as 1/2 (109o 28′ – 60o ) = 24o 44′
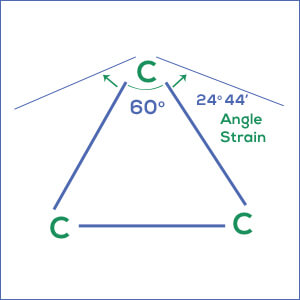
Angle strain in cyclopropane
Advantages of Baeyer’s strain theory:
- Cyclopropane, cyclobutane are more reactive as they have higher angle strain.
- Angle strain in cyclopentane is minimum. Hence, it is most stable.
- The relative stabilities of cycloalkanes up to cyclopentane can be explained satisfactorily.
Limitations of Baeyer’s strain theory:
- According to the theory, cyclohexane is less stable than cyclopentane. However, cyclohexane and other higher cycloalkanes are found to be more stable than cyclopentane.
- Planar model cycloalkanes are explained in the theory.
- Carbon-carbon double bond is easily formed. But according to Baeyer’s strain theory it is difficult to form
Energy diagram of n-Butane
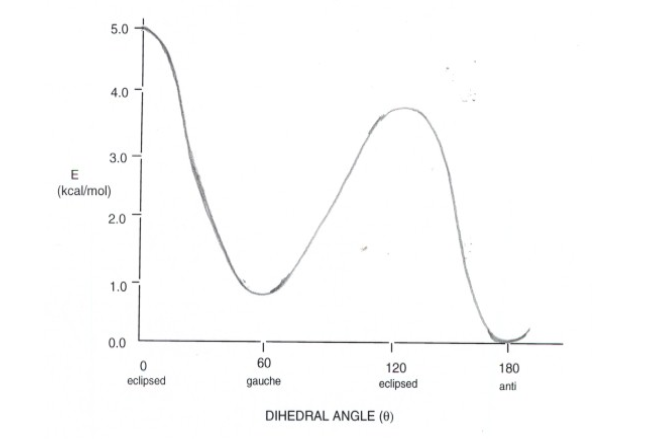
- Torsional strain is absent as both the gauche and anti forms are staggered, which are the two energy maxima.
- The minimum absolute energy is the anti-form as the gauche form has small steric interaction between the two methyl groups. At a dihedral angle of 60 degrees, one hydrogen of each of the methyl groups is relatively close to a hydrogen of the other methyl group (van der Waals repulsion). When we observe carefully observe the steric strain and torsional strain. The latter arises from eclipsed bonds, while the former arises from atoms which are too close to each other.
- There is also two maxima energy present, as both are eclipsed, they are torsionally strained, however, the higher energy conformations have steric strain.
Conformation of n-butane
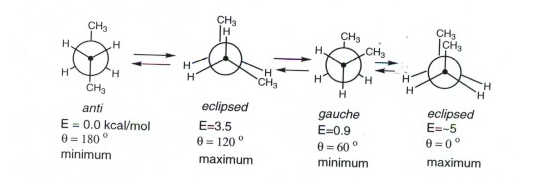
4 carbons, atoms are present in the organic compound Butane, it is also a mixture of 3 isomers. Butane is a gas under atmospheric pressure, they are liquefied gas that is highly flammable.
Compared to ethane, hydrogen butane consists of the complex set of conformations that is related to the constitution. The above figure represents the 4 conformations of butane.
The above diagram explains the rotation about C2-C3 bond due to the change in potential energy.
Conformations of Ethane
Ethane is an organic Chemical Compound. Ethane is a colourless gas and also odourless at standard temperature, they consist of seven sigma bonds. The shape of the molecule changes when a rotation of the six carbon-hydrogen bond occurs. But many possible differences occur when there is a rotation about the carbon-carbon bond.
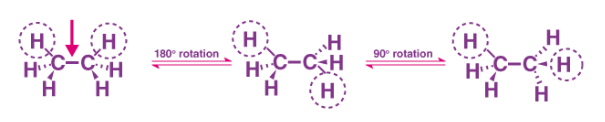
Now suppose we rotate CH3 group clockwise at an angle of 60 degrees, there would be a possible that hydrogen present at the front carbon is close to the hydrogen present at the back carbon. That is Eclipsed Conformation.

Eclipse Conformation is one of the highest Conformation. Another clockwise rotation at an angle of 60 degrees would lead to second eclipsed conformation. The solid line in the above figure represents the 6 carbon-hydrogen bond that is extended at an angle of 120 degrees from 2 carbons
Energy diagram Of Ethane
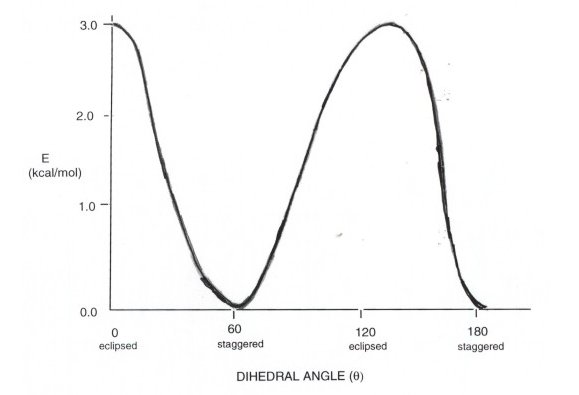
- As rotation around the C-C bond in ethane is continuous, a number of conformations are formed, but the two most important ones are. These are the lowest energy structure, called the staggered conformation, and the highest energy structure, called the eclipsed conformation.
- The staggered structure is important because, being the structure of lowest energy, it is the structure actually adopted by ethane (the ground state structure). It defines the shape of ethane molecules. The eclipsed structure is important not because it is highly populated, but because its energy relative to the staggered structure defines how much energy is required to complete a rotation in ethane. This is called the energy barrier to rotation, and it is about 3 kcal/mol. That is, the eclipsed structure is 3 kcal higher in energy than the staggered structure.
Conformation of Cyclohexane
The most widely occurring ring compound that is natural in its origin, is the Cyclohexane is the most widely occurring ring in compounds of natural origin. Its prevalence, as a result of its stability it is one of the most important cycloalkanes undoubtedly. However, the deviation of bond angle cyclohexane is more than cyclopentane molecule., it should be more strained and less reactive than cyclopentane. But actually, it is less strained and more stable than cyclopentane.
As expected, cyclohexane does not exist as a planar molecule to avoid stain. It exists as a puckered ring which is non-planar and the bond angles are close to tetrahedral bond angles. Two such puckered rings for cyclohexane called boat and chair conformations.
Conformation of Cyclohexane Stability
Generally, in the chair shaped conformation of cyclohexane, there are two carbon-hydrogen bonds of each of the following types:
- Axial ‘up’
- Axial ‘down’
- Equatorial ‘up’
- Equatorial ‘down’
This geometry of chair cyclohexane conformations is generally preserved when the hydrogen atoms are replaced by halogen atoms such as fluorine, chlorine, bromine, and iodine. The phenomenon wherein the cyclohexane molecule undergoes a conversion from one chair form to a different chair form is called chair flipping (or ring flipping).
An illustration detailing chair flipping is provided below.
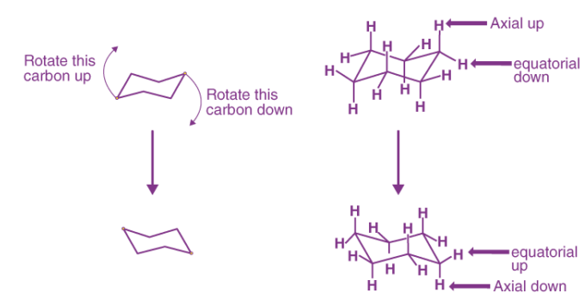
When chair flipping occurs, axial carbon-hydrogen bonds become equatorial and the equatorial carbon-hydrogen bonds become axial. However, they retain the corresponding ‘up’ or ‘down’ positions.
It can be noted that at a temperature of 25o Celsius, 99.99% of the molecules belonging to a given cyclohexane solution would correspond to a chair-type conformation.
The boat conformation of cyclohexane is not a very stable form due to the torsional strain applied to the cyclohexane molecule. The stability of this form is further affected by steric interactions between the hydrogen atoms. Owing to these factors, these conformations are generally converted into twist-boat forms which have a lower torsional strain and steric strain in them.
These twist-boat conformations of cyclohexane are much more stable than their boat-shaped counterparts. This conformation has a concentration of less than 1% in a solution of cyclohexane at 25o. In order to increase the concentration of this conformation, the cyclohexane solution must be heated to 1073K and then cooled to 40K
Key takeaway
The simplest organic compounds are hydrocarbons and are composed of carbon and hydrogen.
Hydrocarbons can be aliphatic or aromatic; aliphatic hydrocarbons are divided into alkanes, alkenes, and alkynes. The combustion of hydrocarbons is a primary source of energy for our society.
There are three types of aliphatic hydrocarbons.
Alkanes are aliphatic hydrocarbons with only single covalent bonds.
Alkenes are hydrocarbons that contain at least one C-C double bond, and alkynes are hydrocarbons that contain a C-C triple bond.
References:
1. Graham Solomons T. W., Fryhle, Craig B., Snyder Scott A, Organic Chemistry, Wiley Student Ed, 11th Edition (2013)
2. Jonathan Clayden, Nick Greeves, Stuart Warren, Organic Chemistry, 2nd Edition, Oxford Publisher, 2014.
3. Dhawan, S.N., Pradeep’s Organic Chemistry, (Vol. I and II), Pradeep Publications




