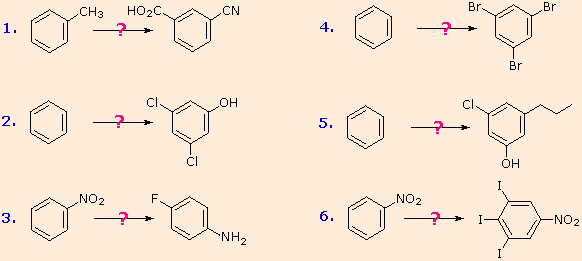
Unit - 1
Nitrogen Containing Functional Groups
1.1.1 Preparation of Amines
Amines are prepared by the following methods:
Nitro compound reduction
Alkyl halide ammonolysis
Nitrile reduction
Amino acid reduction
phthalimide synthesis by Gabriel
Degradation of Hoffmann bromamide
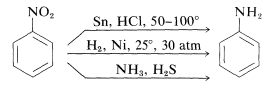
Reduction of nitro compounds
By moving hydrogen gas through finely divided nickel, palladium, or platinum, nitro compounds are reduced to amines, as well as by reduction with metals in an acidic medium.
R- NO2 + 3H2 à R-NH2 + 2H2O {In the presence of Ni, Pt or Pd}
Ar- NO + 3H2à Ar- NH2 + 2H2O
Nitro alkanes can also be reduced to the corresponding alkanamines in the same way.
Reduction with iron scrap and hydrochloric acid is favoured because the FeCl2 produced during the reaction is hydrolyzed, releasing hydrochloric acid, requiring only a small amount of hydrochloric acid to start the reaction.
Nitro compounds can also be reduced with active metals like Fe, Sn, Zn, and others using concentrated HCl.
R- NO2 + 3H2 à R- NH2 + 2H2O {In the presence of Sn/HCl}
Ar- NO2 + 3H2 à Ar- NH2 + 2H2O
Ammonolysis of alkyl halides
In alkyl or benzyl halides, the carbon-halogen bond can be easily broken.
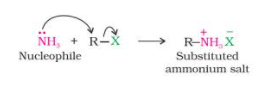
As a result, when an alkyl or benzyl halide reacts with an ethanolic solution of ammonia, the halogen atom is substituted by an amino (–NH2) group in a nucleophilic substitution reaction.
Ammonolysis refers to the cleavage of the C–X bond by an ammonia molecule.
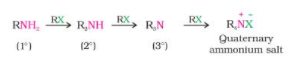
At 373 K, the reaction is carried out in a sealed tube. The resulting primary amine acts as a nucleophile, allowing it to react with alkyl halides to produce secondary and tertiary amines, as well as the quaternary ammonium salt.
Treatment with a solid base yields the free amine from the ammonium salt:
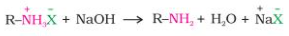
Cons: When ammonia is consumed in excess, primary amine is formed as the main product.
As alkyl halide is used in excess, the main product is quaternary ammonium salt.
Note: This method is not appropriate for the preparation of aryl amines because aryl amines are less reactive towards nucleophilic substitution reactions than alkyl halides.
Reduction of nitriles
Primary amines are formed when nitriles are reduced with lithium aluminium hydride (LiAlH4) or hydrogenated catalytically.
This reaction is used to ascent the amine sequence, that is, to make amines with one more carbon atom than the starting amine.
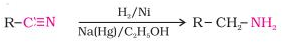
Reduction of amides
When amides are reduced with lithium aluminium hydride, amines are formed.
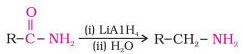
Gabriel phthalimide synthesis
Primary amines are made using the Gabriel synthesis method.
As phthalimide is treated with ethanolic potassium hydroxide, it forms a potassium salt, which when heated with an alkyl halide and then alkaline hydrolyzed yields the primary amine.
Since aryl halides do not undergo nucleophilic substitution with the anion formed by phthalimide, this method cannot be used to make aromatic primary amines.
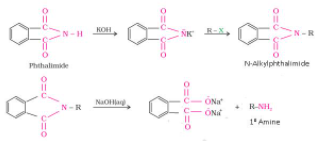
Hoffmann bromamide degradation reaction
Hoffmann developed a process for preparing primary amines by treating an amide with bromine in an aqueous or ethanolic solution of sodium hydroxide. In this degradation reaction, an alkyl or aryl group migrates from the amide's carbonyl carbon to the nitrogen atom, resulting in an amine with one carbon less than the amide.
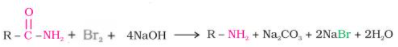
1.1.2 Physical and Spectroscopic Properties:
Nitro compounds are a form of nitrogen derivative that is very essential. Like the carboxylate anion, the nitro group, NO2, is a combination of two equivalent resonance structures:
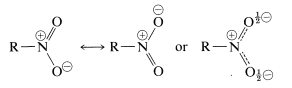
The nitrogen in the hybrid structure has a complete positive charge, while each oxygen has a half-negative charge. This is consistent with nitro compounds' high dipole moments, which range from 3.5D to 4.0D depending on the type of R. Because of the polar character of the nitro group, nitro compounds are less volatile than ketones of similar molecular weight; for example, nitromethane (MW 61) has a boiling point of 101o, while 2-propanone (MW 58) has a boiling point of 56o. Surprisingly, 2-propanone has a poor water solubility; a saturated solution of nitromethane in water is less than 10% by weight, whereas nitromethane is entirely miscible with water.
Powerful infrared bands at around 1550cm1 and 1375cm1 distinguish nitroalkanes from aromatic nitro compounds, while the corresponding bands in the spectra of aromatic nitro compounds occur at slightly lower frequencies. Aromatic nitro compounds, such as nitrobenzene, have extended conjugation and absorb at longer wavelengths (330nm), while nitroalkanes have a weak n transition in their electronic spectra around 270nm.
1.1.3 Preparation of Nitro Compounds:
Nitro compounds can be made in a variety of ways, including by replacing hydrocarbons with nitric acid directly.
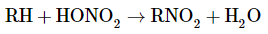
by nitrite ion displacement reactions,
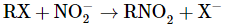
as well as the degradation of primary amines
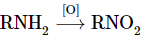
Alkane nitration is only effective when done at high temperatures in the vapour phase. Inevitably, product mixtures are created:
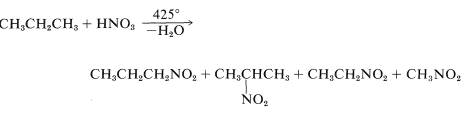
Direct nitration of aromatic compounds such as benzene, on the other hand, occurs readily in the liquid form.
Nitration of a substituted benzene with an electron withdrawing substituent (NO2, CO2H, CN) creates the 1,3-isomer, as it does with other electrophilic substitutions. Less direct routes are needed to prepare the 1,4-isomer, with the most common strategy being to use benzene derivatives with substituent groups that produce the desired orientation on nitration, followed by the requisite modifications in these groups to produce the final product. Thus, 1,4-dinitrobenzene cannot be made from nitrobenzene by nitration, but it can be made from benzenamine using the procedure shown in Figure 24-5. N-phenylethanamide (acetanilide) is formed from benzenamine, and nitration produces the 1,4-isomer. 1,4-dinitrobenzene is generated by hydrolyzing the amide to 4-nitrobenzenamine and replacing the amino with nitro using the nitrite ion in the presence of cuprous salts. Alternatively, trifluoroperoxyacetic acid can oxidise the amino group of 4-nitrobenzenamine to a nitro group. N-phenylethanamide is nitrated instead of benzenamine in these syntheses because benzenamine is readily oxidised by nitric acid, and the nitration reaction results in extensive 3-substitution due to the formation of phenylammonium ion. Another way to make 4-nitrobenzenamine is to nitrate chlorobenzene and then substitute the chlorine with ammonia in a reaction. The nitrations described above produce mixtures of 2- and 4-isomers, which are typically simple to separate using distillation or crystallisation. 4-nitrobenzoic acid can be synthesised using the same process. The methyl group in methylbenzene preferentially guides nitration to the 4 positions, yielding 4-nitrobenzoic acid after oxidation with chromic acid:
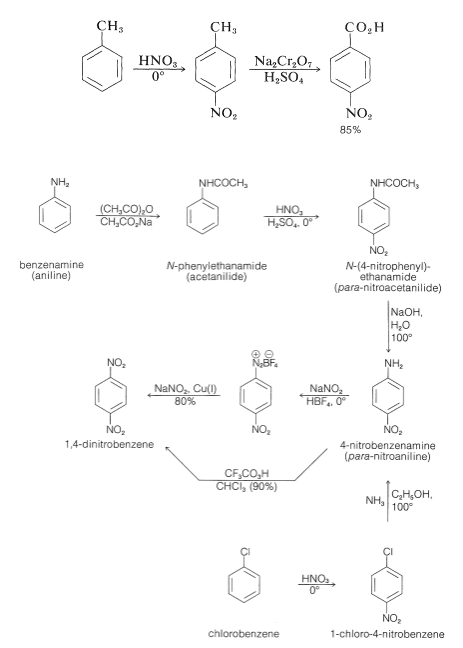
Figure: Synthetic routes to 1,4-dinitrobenzene.
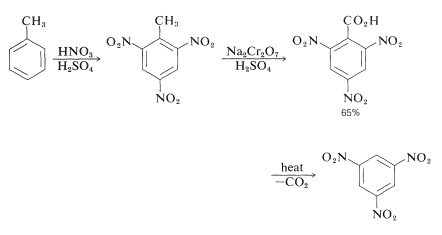
In certain situations, an activating party might be required to encourage replacement, which would otherwise be extremely difficult. The preparation of 1,3,5-trinitrobenzene is a good example; direct substitution of 1,3-dinitrobenzene in fuming sulfuric acid necessitates long heating with nitric acid. However, methylbenzene readily converts to the trinitro derivative, which when oxidised and decarboxylated yields 1,3,5-trinitrobenzene:
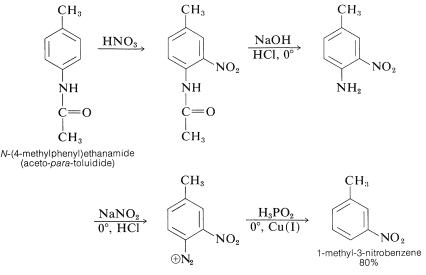
Acylamino groups are also useful activating groups since the amino groups obtained after acyl function hydrolysis can be extracted from an aromatic ring by reducing the corresponding diazonium salt with hypo phosphorous acid, ideally in the presence of copper (I) ions. The following is an example of how 1-methyl-3-nitrobenzene can be made from N-(4-methylphenyl) ethanamide (aceto-para-toluidide):
The reaction of an alkyl halide (with strong SN2 reactivity) with the nitrite ion is one route to aliphatic nitro compounds. Methylsulfinylmethane [dimethyl sulfoxide, (CH3)2SO] and dimethyl methanamide are good solvents (dimethylformamide). As can be seen from Equation 24-6, formation of the nitrite ester by O- instead of N-alkylation is a competing reaction:
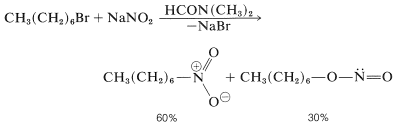
In certain cases, silver nitrite is preferred over sodium nitrite, and the solvent is normally diethyl ether:
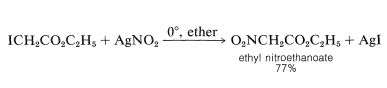
The reaction can be used to make nitromethane quickly.
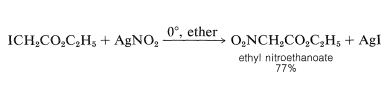
With aryl halides, displacement reactions with the nitrite ion do not perform well. Displacement of the diazonium group, on the other hand, is a realistic route to nitroarenes (the Sandmeyer reaction).
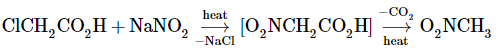
1.1.4 Reactions of Nitro Compounds:
In thermodynamic terms, nitro compounds are very unstable; for example, the heat of decomposition of nitromethane is 67.4kcal mol1 according to the following stoichiometry.
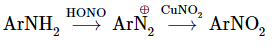
In the industrial use of nitro compounds as explosives, the significant energies and rapid rates of reactions such as this are exploited. There is also the advantage of low shock sensitivity for certain nitro compounds, such as TNT.
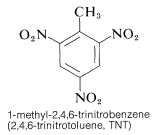
TNT is difficult to detonate by simple contact, and it can also burn without exploding. Decomposition, on the other hand, spreads quickly until detonation begins. Nitro compounds are especially useful due to their fair handling stability and high thermodynamic potential. PETN, cyclonite, picric acid, and tetryl are other polynitro compounds that can be used as explosives:
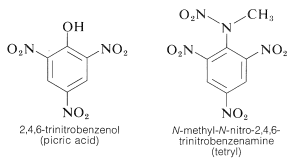
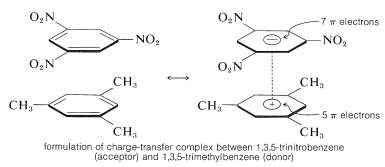
The ability of aromatic polynitro compounds to form "charge-transfer" complexes with aromatic hydrocarbons, especially those substituted with alkyl groups, is a key feature. Complexes of 2,4,6-trinitrobenzenol (picric acid) and aromatic hydrocarbons are often crystalline solids that can be used for aromatic hydrocarbon isolation, purification, and identification. These compounds are known as "hydrocarbon picrates," but this is a misnomer since they are not salts. Additionally, similar complexes form between aromatic hydrocarbons and trinitrobenzene, demonstrating that the nitro groups, not the hydroxyl groups, are needed for complex formation. The binding in these complexes is similar to that found in halogen-alkene-benzene complexes and is caused by attractive forces between electron-rich and electron-poor substances. The term "charge-transfer complex" implies that the complex has VB structures that include an electron transfer from the donor (electron-rich) molecule to the acceptor (electron-poor) molecule. The term "complex" is often used because at least one of the complex's components has a -electron framework. Sandwich-type structures tend to result from charge-transfer or complexes between polynitro compounds and aromatic hydrocarbons, with the aromatic rings in parallel planes but not generally centred exactly over one another:
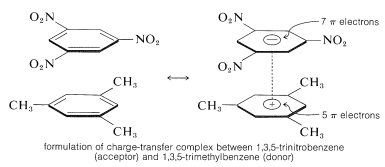
Almost always, charge-transfer complexes are more colourful than their constituents. A spectacular example is benzene and tetracyanoethene, which are both colourless on their own but combine to form a bright-orange complex. Because of the increased probability of stabilising the excited state by electron delocalization involving both components, charge-transfer complexes should exhibit a change toward longer wavelengths of absorption in comparison to their components.
Nitro compounds are easily reduced with a variety of reducing agents, and these reductions allow for the synthesis of aromatic amines:
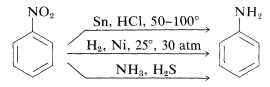
Six equivalents of reducing agent are needed to convert a nitro compound to an amine:
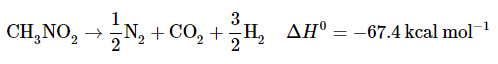
Such a reduction will not be expected to happen in a single move. Indeed, reduction is stepwise and occurs through a series of intermediates that, in the presence of strong reducing agents in acid solution, only exist for a short time. Nitroso compounds, RN=O, and N-substituted azanols (hydroxylamines), RNHOH, are intermediates formed sequentially from RNO2RNO2 by increments of two equivalents of reducing agent:
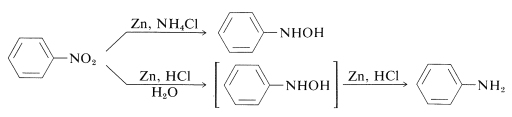
With zinc and ammonium chloride solution, N-aryl-substituted azanols can be derived directly from the corresponding nitro compounds. Zinc and hydrochloric acid, on the other hand, produce the amine:
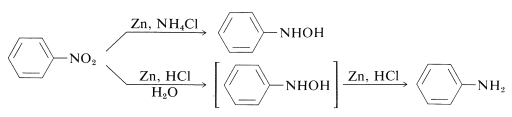
The rate of reduction associated with the acidity of the solution differs between these reactions. The pH of ammonium chloride solutions is around 6. Ammonium chloride is a much weaker acid than HCl.
Nitroso compounds are formed when N-arylazanols are oxidised under controlled conditions. The oxidation of alcohols to ketones is similar to this reaction.
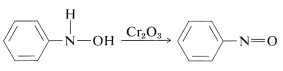
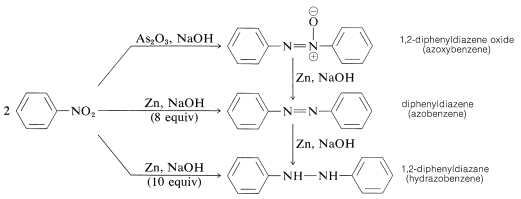
When aryl nitro compounds are reduced with less powerful reducing agents, particularly in alkaline media, a mysterious conglomerate of bimolecular reduction products appears. When it comes to nitrobenzene, for example,
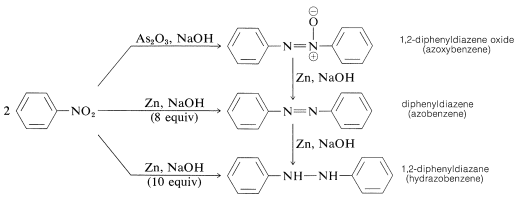
With tin and hydrochloric acid, both of these compounds can be reduced to benzenamine. As a consequence, each may be an intermediate in the reduction of nitro compounds to amines, but this is not always the case. Base-induced reactions between nitroso compounds and azanols or amines create bimolecular reduction products, which may be followed by further reduction of the initially formed substances.
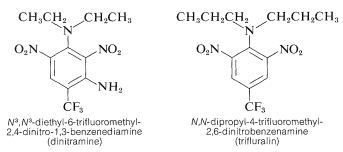
A number of polynitrobenzene derivatives are effective herbicides. N3, N3-diethyl-6-trifluoromethyl-2,4-dinitro-1,3-benzenediamine and N, N-dipropyl-4-trifluoromethyl-2,6-dinitrobenzenediamine are two examples of N3, N3-diethyl-6-trifluoromethyl-2,4-dinitro-1,3-benzenediamine:
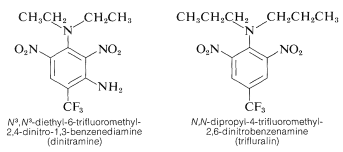
When combined with soil, these chemicals destroy weed seedlings but not crop plants like cotton, soybeans, or peanuts. The behaviour is high; for effective weed control, only 0.08g m20.08g is needed.
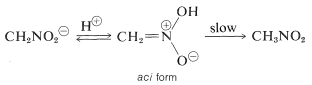
The reactions involving the hydrogens of the primary and secondary compounds are the most critical for nitroalkanes. Nitromethane, for example, is acidic enough to dissolve in aqueous hydroxide solutions. The resulting anion has an electronic structure similar to that of the nitrate anion:
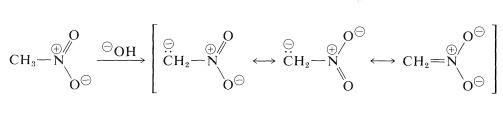
When this ion's solutions are acidified, an unstable, very strongly acidic isomer of nitromethane (referred to as the aci form) is formed, which slowly reverts to the more stable nitro form:
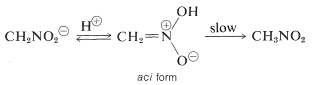
The acidification of a carbonyl compound's enol salt occurs in a similar way, with the only difference being that the aci-nitro compound has a much longer life than an enol of a simple ketone.
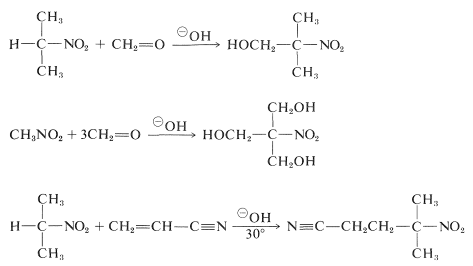
With appropriate carbonyl compounds and simple catalysts, primary and secondary nitro compounds undergo aldol and Michael additions:
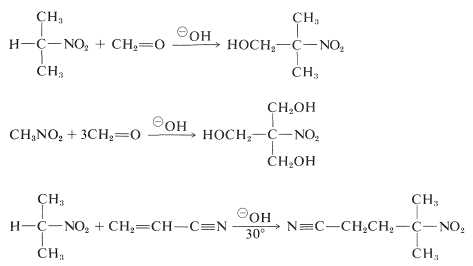
Unfortunately, since CC-alkylation of the conjugate bases of primary nitro compounds is slower than OO-alkylation, alkylation reactions similar to base-catalyzed alkylation of carbonyl compounds are not useful for the synthesis of higher nitro compounds.
1.1.5 Distinguish test between nitroalkanes and alkyl nitrites:
1. Nitroalkane on reduction with H2/Ni produce 1o amines while alkyl nitrites produce alcohols and NH3
Ethyl nitrite
2.Nitroalkanes do not get hydrolysed in basic conditions while nitrites produce alcohols
1.1.6 Cyanides and Isocyanides
Both alkyl cyanides (RCN) and alkyl isocyanides (RNC) are organic derivatives of hydrocyanic acid HCN. Alkali cyanides are ionic 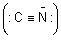 and cyanide ion is ambident in nature (can form covalent bond either from carbon or nitrogen). AgC = N is covalent, hence lone pair on nitrogen is mainly available for covalent bond formation, resulting in predominant formation of isocyanides.
and cyanide ion is ambident in nature (can form covalent bond either from carbon or nitrogen). AgC = N is covalent, hence lone pair on nitrogen is mainly available for covalent bond formation, resulting in predominant formation of isocyanides.
Illustration. How would you account for the fact that alkyl cyanides are soluble in water but alkyl isocyanides are insoluble in water?
Solution: Alkyl cyanides possess the tendency to form H – bonding with water which is absent with isocyanides
1.1.7 Methods of preparation of Cyanides
1. Dehydration of Amides:
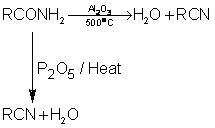
High molecular weight acid amides are dehydrated to the corresponding cyanide by heat alone.
CH3(CH3)6 OCNH2  CH3(CH2)6 CN
CH3(CH2)6 CN
2. From RX:
RX + KCN ——> RCN + KX
This method is satisfactory only if R is 1o or 2o group. If it is 3o group, then it is converted into alkene.
CH3CH2Cl + KCN → CH3CH2CN + KCl
3. By Grignard’s reagent and Cyanogen chloride reaction:
RMgCl + CICIN → RCN + MgCl2
This is best method for preparing 3o alkyl cyanides.
(CH3)3CMgCl + CICN → (CH3)3 CCN + MgCl2
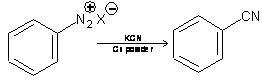
4. From Diazonium salt
1.1.8 Methods of Preparation of Isocyanides
1. By heating an alkyl iodide with AgCN in aqueous ethanolic solution
Rl + AgCN → RNC + Agl
C2H5l + AgCN → C2H5NC + Agl
Ethylisocyanide
2. By carbylamine reaction
Heating a mixture of 1o amine and chloroform with ethanolic potassium hydroxide RNH2 + CHCl2 + 4KOH ——> RNC + 3KCl + 3H2O
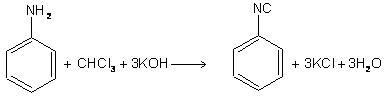
The mechanism works by forming dichloromethylene or dichloro carbene from chloroform in an alkaline solution as an intermediate. (Through a-elimination)
CHCl3 + KOH ———>KCl + H2O +: CCl2
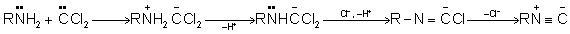
1.1.9 Properties of Isocyanides
1. Alkyl isocyanides are toxic, have a foul odour, and have lower boiling points than their isomeric counterparts.
2. RNC are poorly soluble in water due to the nitrogen atom's lack of a lone pair of electrons for hydrogen bonding.
Reactions:
1. Hydrolysis:
RNC + 2H2O  RNH2 + HCO2H
RNH2 + HCO2H
CH3NC + 2H2O  CH3NH2 + HCO2H
CH3NH2 + HCO2H
RNC are not hydrolysed by alkalis.
2. Reduction:
RNC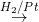 R NHCH3
R NHCH3
2o amine
CH3NC 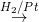 CH3NHCH3
CH3NHCH3
Methyl isocyanide Dimethyl amine
3. When alkyl isocyanides are heated for a long time, they arrange to form cyanide
RNC → R CN
CH3CH2NC → CH3CH2CN
4. With non metals:
(i) RNC + X2 ———> RNCX2
CH3NC + Cl2 ——> CH3NCCl2
(ii) RNC + S ———> RNCS
Alkyl isothiocyanates
CH3NC + S ———> CH3NCS
5. Oxidation with HgO:
RNC + HgO → RNCO + Hg
Akyl isocyanates
CH3NC + HgO → CH3NCO + Hg
Key takeaway:
Nitro compounds are commonly used as a chemical feedstock as well as in the production of medicines, dyes, perfumes, anticancer medications, fertilisers, plastics, and explosives [107,108].
Organic compounds with one or more nitro functional groups (NO2) are known as nitro compounds. The nitro group is one of the most widely used explosophores (functional groups that combine to form a compound explosive). In addition, the nitro group is a heavy electron-withdrawing group.
Metal salts including stannous chloride or chromium (II) chloride are commonly used to convert nitro compounds to oximes. Oximes can also be generated by catalytic hydrogenation with a regulated amount of hydrogen.
Amines are aliphatic and aromatic ammonia compounds. Amines (K b = 10 4) and ammonia (K b = 10 6) are also weak bases. The unshared electron pair on the nitrogen atom causes this basicity.
1.2.1 Classification and nomenclature of amines:
The number of carbon-containing groups attached to the nitrogen atom determines whether amines are listed as principal, secondary, or tertiary. Primary amine compounds have just one group attached to the nitrogen atom, while secondary and tertiary amine compounds have two or three groups attached to the nitrogen atom, respectively.
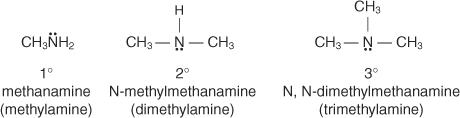
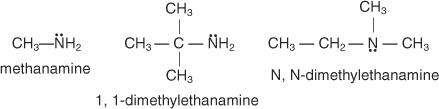
In the common system, you name amines by naming the group or groups attached to the nitrogen atom and adding the word amine.
To call amines in the IUPAC System, use the following rules:
1. Identify the longest continuous carbon atom chain. The alkane with the same number of carbons is the parent’s name.
2. Change the alkane's e to "amine."
3. Identify and label any substituents, bearing in mind that the chain is numbered in the opposite direction of the amine group. A capital N is used to designate substitutes that are bound to the nitrogen atom rather than the carbon of the chain.
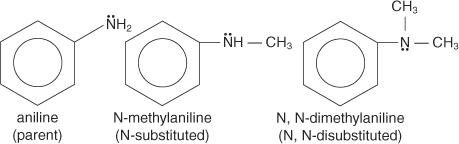
Aromatic amines are divided into several families, each of which acts as a parent molecule. An amino group (—NH 2) attached to benzene, for example, produces aniline as the parent compound.
1.2.2 Basicity of amines:
Amines are classified as basic because they have two unshared electrons that they can share with other atoms. The electron density around the nitrogen atom is produced by these unshared electrons. The more electron density there is in a molecule, the more fundamental it is. The basicity of amines is increased by groups that donate or supply electrons, while groups that decrease the electron density around the nitrogen decrease the basicity of the molecule. The following is the order of base strength for alkyl halides in the gas phase:
(CH 3) 3 N > (CH 3) 2NH > CH 3NH 2 > NH 3
most least
basic basic
However, in aqueous solutions, the order of basicity changes.
(CH 3) 2 NH > CH 3NH 2 > (CH 3) 3N > NH 3
most least
basic basic
Solvation results cause variations in the basicity order in the gas phase and aqueous solutions. Amines occur as ammonium ions in water solutions.
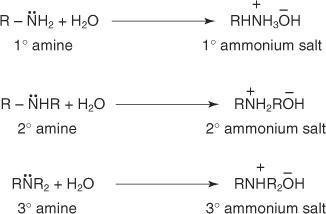
Main and secondary amines' ammonium salts undergo solvation effects (due to hydrogen bonding) in water to a much greater extent than tertiary amines' ammonium salts. The inductive effect of alkyl groups does not increase the electron density on the amine nitrogen as much as these solvation effects do.
Because of resonance, arylamines are weaker bases than cyclohexylamines. Figure 1 shows the resonance structures of aniline, a common arylamine.
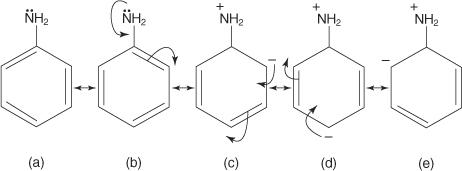
Delocalization of the unshared electron pair occurs in the ring, as seen in structures b through e in Figure, making these electrons less available for reaction. The molecule becomes less fundamental as a result of this electron delocalization.
1.2.3 5 Key Basicity Trends of Amines:
1. Basicity Basics
Fortunately, even if you're just getting started with amines, this topic shouldn't be too unfamiliar to you. You've probably struggled with determining how acidic those molecules are – for example, in the 5 main factors that influence acidity.
You can remember from that unit that something that makes a molecule's conjugate base more stable increases its acidity. [Do you recall Le Châtelier? You'll prefer the equilibrium moving to the right if you make the commodity more stable].
Each of the "factors that increase acidity" has a tendency to stabilise negative charge, either by inductive effects, resonance delocalization, or bringing the charge closer to the nucleus.
Since acidity and basicity are two sides of the same coin, the main factors that influence acidity also influence amine basicity. [Notation 1] As a result, determining basicity entails applying the same ideas in the opposite direction.
In general, the more primitive an electron pair is, the more unstable it is. By eliminating inductive effects, removing delocalization by resonance, or taking the charge farther away from the nucleus, one could increase basicity using the same principles described above.
Let's take a look at each of the main factors one by one to see how they affect the basicity of amines.
2. Basicity Trend #1: Basicity Increases with Increasing Negative Charge on Nitrogen
This is probably the easiest aspect to assess. If "basicity" can be loosely interpreted as "electron-pair instability," and instability rises with charge density, basicity should rise as negative charge rises.
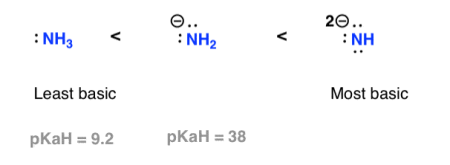
To put it another way, an amine's conjugate base is always a stronger base than the amine itself.
Compare ammonia (NH3) to the amide anion (NH2), which is its conjugate base (-). The amide anion is a much more powerful foundation (pKaH of 38, versus pKaH of 9). It can, for example, deprotonate terminal alkynes (pKa = 25), whereas ammonia cannot.
Effect of charge
Basicity increases with increased negative charge.
3. Basicity Trend #2: “Resonance”, or, Conjugated vs. Non-Conjugated Amines
Lower charge densities lead to lower basicity, and this relationship holds true for lone pairs that can be delocalized into a larger pi system through resonance.
It's worth noting that phenol (pKa = 10) is a far stronger acid than cyclohexanol (pKa = 16).
Since "the heavier the acid, the lower the conjugate base," phenol's conjugate base is weaker than cyclohexanol's conjugate base.
What is the reason for this? Since the conjugate base of phenol can be stabilised by resonance, while the conjugate base of cyclohexanol cannot, this is valid.
The oxygen in phenol is part of a broader "pi scheme," and the electron density in the aromatic ring can be spread by resonance. (Keep in mind that a lower charge density equals more stability.)
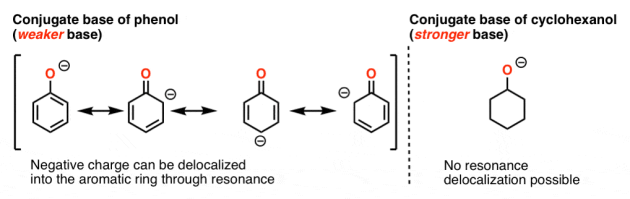
Let's take amines as an example.
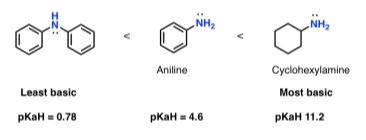
We should also expect aminobenzene (“aniline”) to be a weaker base than cyclohexylamine by comparison.
Yes, that is right! Aniline has a pKaH of 4.6, while cyclohexylamine has a pKaH of 11.2. (The stronger the foundation, the higher the pKaH.)
When a second phenyl ring is added to the nitrogen, the basicity is further reduced (pKaH = 0.78).
Resonance-stabilized amines are less basic
In comparison to non-conjugated amines, lone pairs of amino acids that are conjugated with pi bonds (and thus can participate in resonance) are stabilised.
Basicity is reduced as a result of resonance stabilisation.
The bottom line is that a conjugated amine would be less simple than a non-conjugated amine if all other factors are equal.
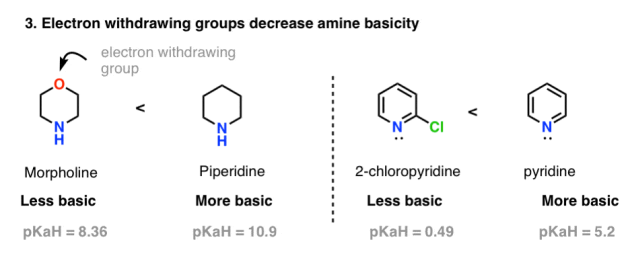
4. Basicity Trend #3. Inductive Effects Decrease Basicity
By sucking electron density away from the conjugate base, electron withdrawing atoms (e.g., F or Cl) or functional groups (e.g., NO2) appear to increase acidity. For e.g., trifluoroethanol (pKa = 12.5) is much more acidic than ethanol (pKa = 16). Lower charge density equals more stability, which equates to lower basicity.
As a result, we'd expect electron withdrawing groups on amines to reduce their basicity as well. Yes, they do! Consider the differences between morpholine (pKaH = 8.36) and piperidine (pKaH = 11), as well as 2-chloropyridine (pKaH = 0.49) and pyridine (pKaH = 5.2).
5. Basicity Trend #4: Pi-Acceptors and Pi-Donors
Resonance and inductive effects also appear to reduce basicity (Factor 2), as we've seen (Factor 3).
However, how do you justify why amides have a lower basicity than amines? Is it a case of resonance? Is it because of inductive effects? Is it a combination of the two?

A section on how nitrogen's basicity is influenced by its interactions with other functional groups in a pi-system seems worthwhile.
When nitrogen acts as a pi-donor, its basicity is reduced, and when it acts as a pi-acceptor, its basicity is increased.
6. Nitrogen Is Less Basic When It Is a Pi-Donor
Let's return to our amide example. What makes it less fundamental?
The presence of an electron-withdrawing oxygen, which can strip some of the electron density from nitrogen, is the first element. However, there is a major resonance form in which the nitrogen lone pair forms a new pi bond with carbon (we call this "pi-donation"), causing a pair of electrons to move from the C-O pi bond to the oxygen (we call this "acting as a pi acceptor").
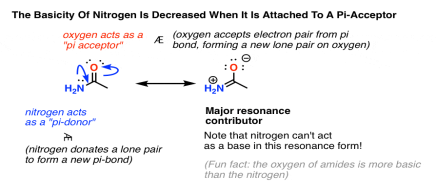
Look at that resonance form on the right. The nitrogen doesn’t have a lone pair anymore, and therefore it cannot act as a base.
Therefore, the basicity of a nitrogen is decreased when attached to a pi-acceptor.
What is pi-acceptors, again? If you’ve covered electrophilic aromatic substitution, these functional groups should seem familiar. You might recognize that “Pi acceptors” all belong in the category of “meta- directors”.

[CF3 is an example of a functional group that is a meta director but not a pi acceptor, since it has no pi bonds]
7. Nitrogen as A Pi-Acceptor
You may wonder if this can also work in the other direction.
When a nitrogen is connected to a pi-donor, will its basicity be increased? Without a doubt.
Pyidine (pKaH = 5.2) and 4-dimethylamino pyridine (pKaH = 5.2) make an excellent contrast (DMAP). When the strongly pi-d0nating NMe2 group is attached to the 4-position, the ring nitrogen's basicity is increased by 104 (pKaH = 9.2). Examining DMAP's resonance forms is instructive. The nitrogen in the ring has a negative charge in the main resonance shape.
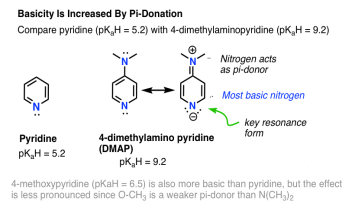
DMAP's ring nitrogen, not NMe2, is the most simple nitrogen! Since NMe2 is a pi-donor (see above), it is made less basic, but the pyridine nitrogen is made more basic because it is the pi-acceptor here.
Guanidine’s are another example of how attachment to pi-donors can increase the basicity of nitrogen. Two pi-donating NH2 groups in guanidine will donate electron density to the (pi-accepting) C=NH.
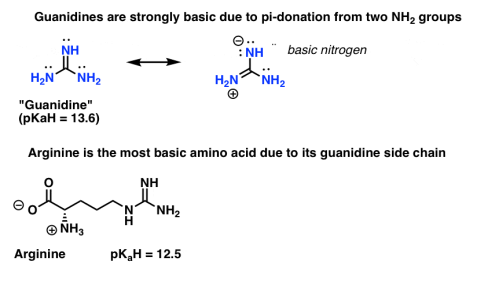
Many who have studied biochemistry may remember that arginine (pKaH = 12.5) is the most basic of the 20 essential amino acids.
8. Basicity Trend#5: Hybridization
One of the more remarkable acidity trends is that alkynes are unusually acidic (pKa = 25) relative to alkenes (pKa’s around 43) and alkanes (pKa’s >50).
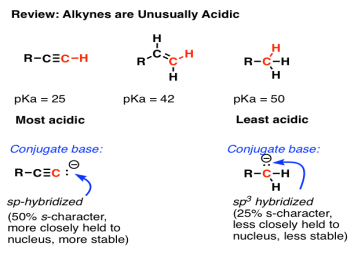
As you might remember, the sp-hybridized orbitals of alkynes have 50% s-character, and since the 2s orbital is closer to the nucleus than the 2p orbitals, the resulting lone pair of the conjugate base "feels" more of the positive charge from the nucleus than a lone pair in a sp3 hybridised orbital (25 percent s-character). It's the same reason why a lone pair on a more electronegative atom like fluorine is more stable than one on a less electronegative atom like carbon.
How would you estimate the relative basicity of nitriles, pyridine, and piperidine if you knew this?
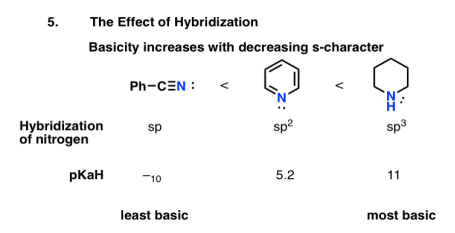
We'd expect the lone pairs in sp-hybridized nitriles to be the most stable and thus the least simple, similar to alkynes. As a result, the lone pairs in sp3-hybridized amines are expected to be the least stable and thus the most fundamental.
The pKaH principles support this. The pKaH of benzonitrile (–10) shows that nitriles are extremely weak bases. The lower basicity of pyridine (pKaH = 5.2) compared to piperidine (pKaH = 11) can also be explained by orbital hybridization.
9. Bonus Factor: Aromaticity
Classic exam question. What’s more basic, pyridine or pyrrole?
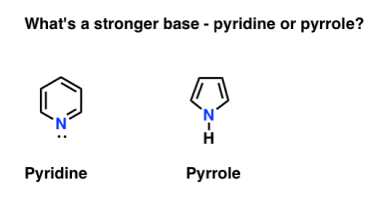
The nitrogen in pyrrole turns out to be unusually non-basic. And when exposed to acid, pyrrole reacts at carbon (C-2) rather than nitrogen (N). Pyridine [pKaH = 5.2] is significantly more fundamental than pyrrole [pKaH = –3.6].
What is the reason for this?
Draw the pyrrole conjugate acid. Have you noticed anything?
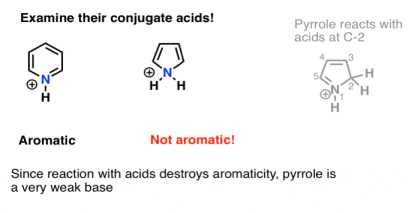
The conjugate acid has no aromatic properties. Protonation of the lone pair on nitrogen will destroy the lone pair's conjugation with the other p orbitals of the ring, rendering the molecule non aromatic.
This can ring a familiar bell. You may recall that cyclopentadiene is an acid that is extremely powerful for a hydrocarbon. “The conjugate base of cyclopentadiene is unusually weak,” we may say again.
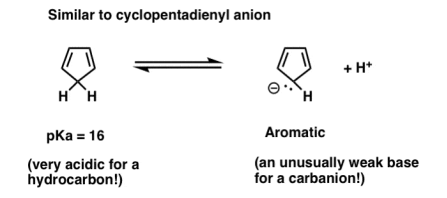
Can you see the pyrrole analogy? Aromaticity is lost when the cyclopentadienyl anion is protonated.
The bottom line is to keep an eye out for situations where forming a new N-H bond could cause aromaticity to be disrupted.
Can you apply the same logic to the following question?
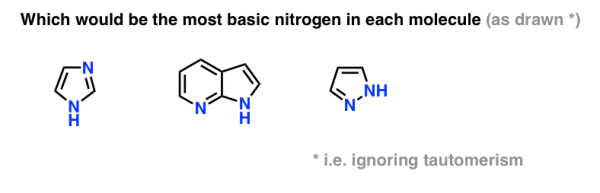
10. Summary: Amine Basicity Trends
Okay, I said there were five factors that influenced acidity, but I actually covered six. The explanation for this is that “pi donation” and “pi accepting” behaviour is rarely discussed in Org 1 (when acidity is introduced), but most students have already experienced these definitions in the form of electrophilic aromatic substitution by the time they encounter amines.
The most important takeaway for today is that because “the stronger the acid, the weaker the conjugate base” and “the weaker the acid, the stronger the conjugate base,” any factor that affects acidity also affects basicity.
By definition, if you know what causes negative charge to stabilise (and thus make an atom less basic), you also know what causes negative charge to destabilise (and therefore make an atom more basic).
How do we deal with circumstances in which many variables are at play? We'll have to depend on experimentation to find out (pKaH). When more than one variable is updated at the same time, it's impossible to forecast patterns.
Observations
Note 1: The atomic aspects of electronegativity and polarizability aren't explored because we're just talking about nitrogen's basicity.
Remember, however, that basicity is inversely related to electronegativity across a row of the periodic table. H3C (-) is more fundamental than H2N (-), which in turn is more fundamental than HO (-), which is more fundamental than F. (-). The more electronegative an element is, the less stable its lone pair is, and thus the higher its basicity.
Another useful pattern is that when you move down a column of the periodic table, basicity decreases. This is due to the fact that when one descends a column of the periodic table, the size of the valence orbitals increases. As a result, the electrons would be "spread out" over a greater region, resulting in a lower charge density. The word "polarizability" is a common way of explaining this.
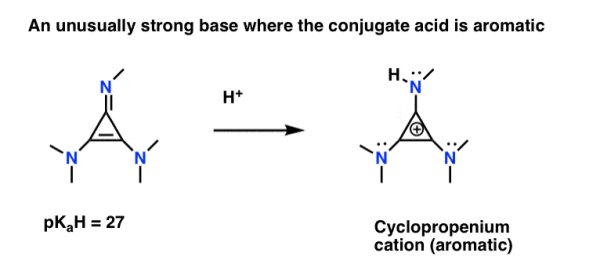
Note 2: Here's a fun example where aromaticity causes nitrogen to be oddly basic. The Columbia University research group of Tristan Lambert has developed a family of imine "superbases."
1.2.4 Structure of Amines
Alkyl Amines
Tetrahedral nitrogen centres make up alkylamines. The bond angle between C-N-C and C-N-H is 109 degrees in this case. As compared to the C-C set, the distance between C-N is shorter. The amines may also be chiral, meaning that the nitrogen centre contains four replacements that form lone pairs.
Aromatic Amines
Aromatic amines (also known as "anilines") have a nearly planar structure of nitrogen. This is due to the aryl substituent mixing with the lone pair. The C-N spectrum is a lot smaller. The difference between C and N in aniline is the same as the distance between C and C.
Example:
H2N –(CH2)6 – NH2 (Hexane -1,6-diamine or 1,6-hexane diamine)
1.2.5 Preparation of Amines and Reactions
The development of amines can be accomplished in a variety of ways. Ammonia reactions with organic compounds are two commonly used processes.
Chemical reduction occurs when the oxygen atom in a molecule is substituted by hydrogen atoms.
Alkylation
Amines are made industrially by alkylation of alcohols with ammonia.
ROH + NH3 RNH2 + H2O ROH + NH3 RNH2 + H2O ROH + NH3 RNH2 + H
Acylation
The reaction between chlorine, anhydride, and an ester is known as acylation. It's a nucleophilic substitution reaction of some sort. The hydrogen atom is replaced with an acyl group in this reaction.
Benzoylation
We will consider the reaction between methenamine and benzoyl chloride which results in the formation of hydrochloric acid and N-methylbenzamide.
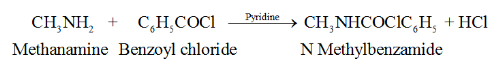
1.2.6 Reduction of Nitro Compounds
The nitro compound can be reduced in one of two ways:
Chemical reduction catalytic hydrogenation catalytic hydrogenation catalytic If the molecule contains another easily hydrogenated group, such as a double bond with carbon dioxide, this method cannot be used. Applying hydrochloric acid to a mixture of nitro compound and metal, normally granulated tin or iron, is the most effective chemical treatment.
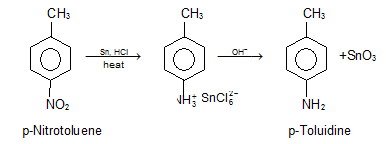
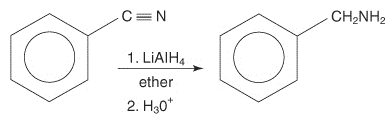
1.2.7 Reduction of Nitriles
Nitriles may be reduced to primary amines by lithium aluminium hydride (LiAIH4).
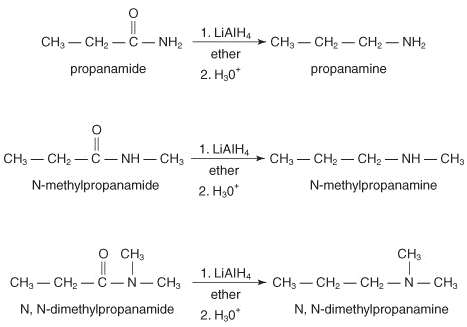
1.2.8 Reduction of Amides
By reducing lithium aluminium hydride, amides produce primary amines, while N-replaced and N-replaced amides produce secondary and tertiary amines, respectively. Since amides are so easy to make, reduction is a popular method for creating all amine groups.
1.2.9 Reductive Amination of Aldehydes and Ketones
In the presence of ammonia or primary or secondary amines, catalytic or chemical reactions may remove aldehydes or ketones, resulting in primary, secondary, or tertiary amines. The reaction of a ketone with ammonia, followed by catalytic reduction or degradation by sodium cyanoborohydride, yields a 1 ° amine.
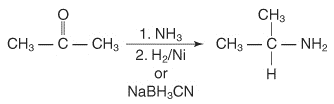
Ketone reactions with primary amines produce N-replaced amines, which are then reduced.
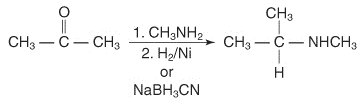
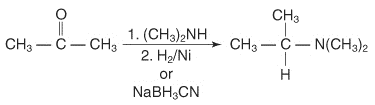
2° amine reaction with ketones followed by reduction can produce N, N-replaced amines.
Commercially, nitrogen is obtained by liquefaction and fractional distillation of air. There are two key phases in this procedure:
Step 1: High pressure between 100 and 200 atmospheres is used to reduce air to liquid air. The compressed air is then expanded by passing it through a fine jet. This process is replicated several times, resulting in liquid air formation.
Step 2: Fractional distillation is performed on the resulting liquid. Dinitrogen has a lower boiling point than liquid oxygen, so it separates from it, leaving liquid oxygen behind. The impure liquid serves as a source of nitrogen.
In laboratory, dinitrogen is obtained by reacting aqueous solution of ammonium chloride with sodium nitrite.
N2(g)+ 2H2O(l) + NaCl = NH4Cl(aq) + NaNO2(aq) (aq)
Impurities such as NO and HNO3 are present in the material, which can be eliminated by thermal decomposition of ammonium dichromate. Passing the gaseous mixture by sulphuric acid containing potassium dichromate is another way to eliminate impurities.
N2+ 4H2O+ Cr2O3 (NH4)2Cr2O7
Pure nitrogen is formed during the decomposition of sodium or barium azide in the presence of high temperature.
1.3.2 Physical properties of Dinitrogen:
In nature, nitrogen is colourless, odourless, and diamagnetic. It's a non-toxic gas that's only slightly soluble in water.
Nitrogen condenses to form a colourless vapour, which then solidifies to form a snow-like mass.
1.3.3 Chemical properties of Dinitrogen:
Because of the N = N bond, dinitrogen has a high bond enthalpy. It is inert at room temperature as a result of this. However, as the temperature rises, the reactivity rises with it. Nitrogen molecules react with metals to form ionic nitrides and non-metals to form covalent nitrides at high temperatures.
2Li3N 6Li +N2heat 6Li +N2heat 6Li +N2heat 6Li +N2heat 6Li +
In Haber's Process, it reacts with hydrogen to form ammonia at 773 K.
773k 2NH3(g) + 3H2(g) N2(g) + 3H2(g) N2(g) + 3H2(g) N2(g) + 3H2(g) N
When a nitrogen molecule interacts with an oxygen molecule at a temperature of 2000 K, nitric oxide is formed.
2NO = N2(g) + O2(g) (g)
1.3.4 Dinitrogen's Applications:
It is primarily used in the industrial production of compounds like ammonia and calcium cyanamide.
It is used to create an inert environment in manufacturing industries such as iron and steel.
Liquid nitrogen is used as a preservative and a refrigerant in the food industry.
1.3.5 PREPARATION OF DIOXYGEN
The catalytic decomposition of solid potassium chlorate is the most popular and practical method for producing dioxygen in the laboratory. The catalyst in this reaction is manganese dioxide.
2KClO3 = 2KCl +3O2 = 2KCl +3O2 = 2KCl +3O2 = 2KCl +3O2 = 2
MnO2 (Manganese Oxide)
The thermal decomposition of metal oxides from the lower part of the electrochemical sequence is another laboratory process. Dioxygen is generated by the thermal decomposition of silver oxide or mercuric oxide, for example.
2Ag2O → 4Ag + O2
Silver oxide Δ Silver Dioxygen
2HgO → 2Hg + O2
Mercuric oxide Δ Mercury Dioxygen
Dioxygen may also be obtained in the laboratory by heating the higher oxides of some metals like lead, barium, and manganese.
2PbO2 → 2PbO + O2
Lead (IV) oxide Δ Lead (II) oxide Dioxygen
2BaO2 → 2BaO + O2
Barium peroxide Δ Barium oxide Dioxygen
2MnO2 + 2H2SO4 → 2MnSO4 + 2H2O + O2
Manganese Sulphuric Δ manganese Water Dioxygen
(IV) oxide acid (II) sulphate
Salts that are rich in oxygen, such as permanganates and nitrates, when decomposed thermally also yields dioxygen.
2KNO3 → 2KNO2 + O2
Potassium Δ Potassium Dioxygen
nitrate nitrite
2KMnO4 → K2MnO4 + MnO2 + O2
Potassium Δ Potassium manganese Dioxygen
permanganate manganate (IV) oxide
2NaNO3 → 2NaNO2 + O2
Sodium Δ Sodium Dioxygen
nitrate nitrite
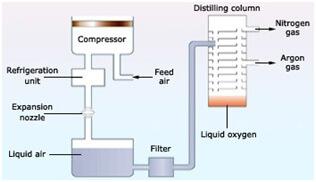
1.3.6 The Fractional Distillation of Liquid Air
The aim of an air separation device, also known as an oxygen or nitrogen generator, is to remove one or both of these elements from the air. The air is first passed through a filter, which absorbs all of the water vapour in this distillation process.
2. The cooling process, which employs turbines and high-energy refrigeration systems, then starts.
3. As the temperature exceeds the sublimation or boiling points of any of the component gases, carbon dioxide and other trace gases settle out. The transformation of matter from a solid to a gaseous state is known as sublimation.
4. When the temperature exceeds 200 degrees Celsius, the liquified mixture is poured into a flask that is slightly colder at the bottom (185 degrees Celsius) than at the top (190 degrees Celsius).
5. Oxygen liquefies at −183 °C, therefore it flows out of the flask through a tube from the bottom of the vessel. The boiling point of nitrogen is −196 °C. It flows out through a tube connected to the top of the flask.
1.3.7 PHYSICAL PROPERTIES OF DIOXYGEN
It's a colourless, odourless, and tasteless gas that's slightly heavier than air and slightly soluble in water. This tiny amount of dissolved dioxygen is only enough to keep marine life alive.
The oxygen gas can be liquefied under pressure to a light blue liquid by compressing it at 90K. At 55K, it can also solidify into a bluish-white solid.
1.3.8 CHEMICAL PROPERTIES OF DIOXYGEN
Dioxygen is a highly reactive gas that reacts with almost all metals and nonmetals directly. Metals like gold and platinum, as well as noble gases like helium, neon, and argon, do not react directly with it.
The reaction of dioxygen with metals
Most metals burn in the presence of oxygen to produce oxides, which are the most essential in nature. Since they react with dilute acids to form salt and water, all metal oxides are basic in nature. They also dissolve in water to form metal hydroxides, which are alkaline in nature. Since these metal hydroxides release OH- ions, they are considered basic.
Metal Dioxygen Metal-oxide
4M + O2 → 2M2O
Δ
2M + O2 → 2MO
Δ
4M + 3O2 → 2M2O3
Δ
Non-metals burn in the presence of dioxygen to form acidic oxides. For example, sulfur burns in the presence of oxygen give sulfur dioxide.
Δ
S + O2 → SO2
Since they react with water to create corresponding acids, non-metallic oxides are acidic in nature. As SO2 reacts with water, it releases sulphuric acid (H2SO4). As a result, SO2 is an acidic gas.
Reactions of dioxygen with some compounds:
Sulfur dioxide is oxidised to sulphur trioxide in the presence of a catalyst, vanadium pentoxide. It's a crucial step in the contact process's production of sulphuric acid.
V2O5
2SO2 + O2 → 2SO3
Dioxygen reacts with several organic compounds, such as hydrocarbons and carbohydrates, at very high temperatures or on the ignition, forming carbon dioxide and water.
High Temperature
CH4 + 2O2 → CO2 + 2H2O
Methane Dioxygen Carbon Water
dioxide
High Temperature
C6H12O6 + 6O2 → 6CO2 + 6H2O
Glucose Dioxygen Carbon Water
dioxide
1.3.9 USES OF DIOXYGEN
Dioxygen is used in the manufacturing of many metals and is used in processes such as combustion and respiration. It is combined with carbon dioxide or helium to be used for artificial respiration.
It is widely used in metal cutting and oxy-acetylene welding.
In the production of nitric acid, dioxygen is used to oxidise ammonia.
It's found in oxygen cylinders, which are common in hospitals, high-altitude flights, and mountaineering.
Rocket fuel contains liquid oxygen, which is an essential component.
1.4.1 What is Gabriel Phthalimide Synthesis Reaction?
There are three stages in the Gabriel Phthalimide Synthesis Mechanism. The Synthesis is named after German scientist Siegmund Gabriel and is used to make primary amines from primary alkyl halides.
The reaction has been generalised for use in the alkylation and deprotection of sulfonamides and imides to produce amines. Since ammonia alkylation is inefficient, it is replaced with phthalimide anion in the Gabriel synthesis.
1.4.2 Synthesis Details
The Gabriel synthesis has the greatest benefit of avoiding over alkylation. The reaction of potassium hydroxide with phthalimide also produces a strong nucleophile in the form of an imide ion. The imide ion performs a nucleophilic substitution reaction on the alkyl halide, yielding N-alkyl phthalimide as an intermediate. This phthalimide is hydrolyzed or hydrazinolyzed to produce a primary alkyl amine. Aryl amines, on the other hand, cannot be made using Gabriel synthesis because aryl halides do not undergo simple nucleophilic substitution.
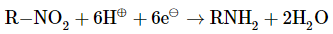
1.4.3 Gabriel Phthalimide Synthesis Mechanism
Step 1: An acid-base reaction occurs when potassium hydroxide is added to the phthalimide. The imide is deprotonated by the hydroxide ion. The resulting proton is more acidic than any simple amine (resonance stabilisation provided by the two adjacent carbonyl-like groups), resulting in a heavy nucleophile – the imide ion.
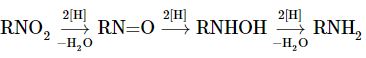
Step 2 The alkyl halide's electrophilic carbon is attacked by the nucleophilic imide ion. The nitrogen atom then bonds with the carbon itself, replacing the halogen (Fluorine, Chlorine, Bromine, or Iodine) in the alkyl halide. An N-Alkyl Phthalimide is formed as a result of this reaction.
3rd phase
The mechanism is very similar to base-catalyzed hydrolysis of esters, but instead of oxygen, nitrogen is added to the R group. The hydroxide ion cleaves the N-Alkyl phthalimide by attacking the carbon atom bound to the nitrogen atom. The oxygen atom is also attached to the cation in the base. As the oxygen atom replaces the nitrogen atom in the phthalimide, the hydrogens expelled from the hydroxide ion bind with the nitrogen atom attached to the R group. Below is an illustration of the third step in the Gabriel phthalimide synthesis mechanism.

Finally, the Gabriel method can be used to synthesise primary amines from phthalimides. Instead of using an aqueous base, acidic hydrolysis or hydrazinolysis may be used to follow the mechanism (as was shown in the mechanism above). With secondary alkyl halides, the Gabriel approach typically fails. Another downside of this synthesis is that it produces a low yield by using acidic/basic hydrolysis, while using hydrazine will make the synthesis conditions relatively harsh.
Key takeaway:
The potassium salt of phthalimide is formed by reacting phthalimide with ethanolic KOH. The N-alkyl phthalimide is then formed by heating it with alkyl halide. A primary amine is generated by alkaline hydrolysis (or hydrazine treatment).
The Synthesis is named after German scientist Siegmund Gabriel and is used to make primary amines from primary alkyl halides. The reaction has been generalised for use in the alkylation and deprotection of sulfonamides and imides to produce amines.
Potassium phthalimide is a -NH2-synthon that can be used to make primary amines when combined with alkyl halides. The phthalimide is no longer a nucleophile and does not react after alkylation. Reaction with base or hydrazine cleaves the product, resulting in a stable cyclic product.
1.5.1 Mechanism:
The addition of amine to the intermediate formed by the dehydrohalogenation of chloroform is part of the carbylamine reaction mechanism. Dichlorocarbene is the name of this intermediate. Hofmann isocyanide synthesis is another name for the carbylamine reaction. Isocyanides are made by combining a primary amine, chloroform, and a base. This conversion relies heavily on the dichlorocarbene intermediate. Isocyanides can't be made from secondary or tertiary amines using the carbylamine reaction. The carbylamine reaction can be written as – in general.
R-NH2 + CHCl3 + 3KOH —→ RNC (Carbylamine) + 3KCl + 3H2O
Here are a few examples of the carbylamine reaction.
1.5.2 Hofmann’s Isocyanide Test
The carbylamine reaction can be used as a chemical test for the existence of primary amines since it is only selective for primary amines. The carbylamine reaction is also known as Hofmann's isocyanide test when used as a test. The test substance is heated with chloroform and alcoholic potassium hydroxide in this procedure. In the presence of primary amine, isocyanide (carbylamine) can form, which is readily identifiable by its foul odour. Since secondary and tertiary amines do not undergo the carbylamine reaction, the Hofmann isocyanide test does not produce a foul odour.
1.5.3 Carbylamine Reaction Mechanism
To make dichlorocarbene intermediate, the first step is to dehydrohalogenate chloroform (remove hydrogen halide from a given substrate). The dichlorocarbene intermediate is a highly reactive intermediate. The electrophilic dichlorocarbene targets the primary amine's nucleophilic nitrogen. Isonitrile is formed when hydrochloric acid is removed from the system. The mechanism of the carbylamine reaction is depicted in the diagram below.
Finally, using chloroform and a base, the carbylamine reaction can be used to make isocyanides from primary amines. The carbylamine reaction can also be used to determine if a substrate contains a primary amine.
Reaction of Mannich
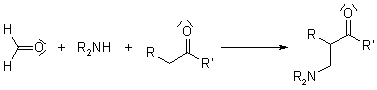
Aminomethylated products are generated by combining a nonenolizable aldehyde, a primary or secondary amine, and an enolizable carbonyl compound in a multi-component condensation. The acceptor in the reaction is the aldehyde's iminium derivative.
Many biosynthetic pathways, especially for alkaloids, have been proposed to include the Mannich Reaction.
1.5.4 Mechanism of the Mannich Reaction
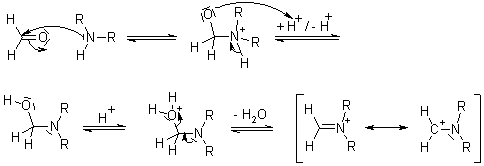
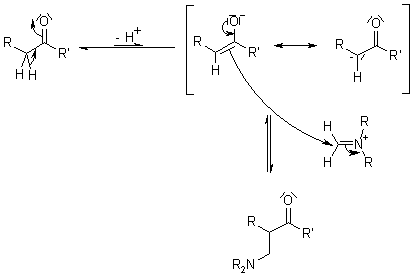
Key takeaway:
The analyte is heated with alcoholic potassium hydroxide and chloroform in this reaction. The isocyanide (carbylamine) is produced when a primary amine is present, as indicated by a foul odour. Secondary and tertiary amines do not respond positively to the carbylamine test.
The response is yes. Carbylamine reaction: i. Aliphatic or aromatic primary amines are converted to foul-smelling alkyl/aryl isocyanides or carbylamines by heating with chloroform. The carbylamine reaction can be used as a chemical test for the existence of primary amines since it is only selective for primary amines. The carbylamine reaction is also known as Hofmann's isocyanide test when used as a test.
1.6.1 The Hofmann Elimination of Alkylammonium Salts: Examples and Mechanism
1. Quick Review: Zaitsev’s Rule
Zaitsev's law governs traditional elimination reactions involving the E2 process. The more substituted alkene would be the main product (that is, the alkene with the most carbons directly attached to the alkene).
The “trisubstituted” alkene is preferred over the “mono-substituted” alkene in the first example below.
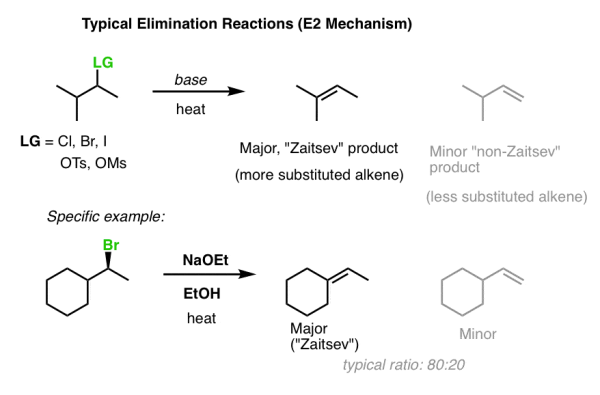
What is the reason for this? Alkenes' thermodynamic stability rises in order of monosubstituted, disubstituted, trisubstituted, and tetrasubstituted.
The energy differences are minor – around 2 kcal/mol – but they are sufficient to produce an 80:20 product ratio! [How did we come to this conclusion? It's calculated by substituting 2 kcal/mol into the equation ln K = –G/RT].
2. “Non-Zaitsev” Products Can Dominate When Sufficient Steric Hindrance Is Present
The use of a bulky base for the elimination reaction may often result in “non-Zaitsev” products. The use of sodium or potassium t-butoxide (KOt-Bu) is a classic example, as is the use of lithium di-isopropyl amide (LDA). The theory is that the bulky base can react more rapidly with the proton on a beta-carbon that is least sterically hindered, resulting in the formation of the least substituted alkene. [See Bulky Bases in Elimination Reactions for more information].
These "non-Zaitsev" products are often referred to as "Hofmann products." What is the reason for this?
Let's get back to amines.
3. The “Hofmann Degradation”
There were few methods for analysing complex molecules available in 1851. Degradation was one technique for determining the composition of an unknown compound by breaking it down into smaller parts and looking for clues in the fragments. August Wilhelm von Hofmann invented a two-step amine degradation process that was later named after him.
The first step is to treat an amine with a large amount of methyl iodide [CH3I], which produces an ammonium salt [we discussed this reaction in a previous article, “exhaustive methylation”).
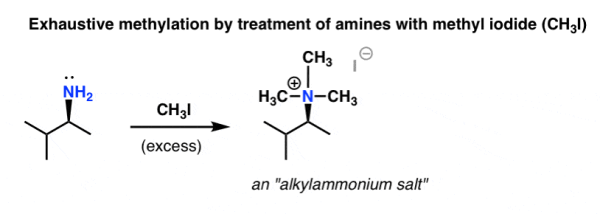
The ammonium salt is then distilled under low pressure in the presence of a strong base in the second step. Silver oxide (Ag2O) is a popular material.
While it may not appear so at first glance, tertiary amines (: NR3) are relatively poor bases (pKaH = 10) and therefore good leaving classes. [Remember that strong bases = good leaving groups]
When heated with a strong base, an elimination reaction occurs, in which NR3 is lost and a new alkene is formed.
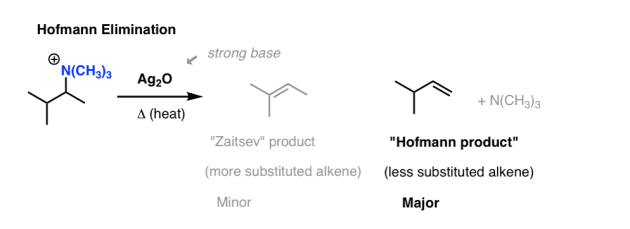
The alkene product from this method appears to be the least substituted alkene (“Hofmann product”) rather than the Zaitsev product, which is an interesting observation.
1. The Hofmann Elimination Has an Extremely Bulky Leaving Group, And This Leads to “Non-Zaitsev” Elimination Products
It's not that the product alkene has a property that makes it more stable than the Zaitsev product (it doesn't).
The relative energies of the transition states that lead to the two products provide the response.
It may be beneficial to revisit the reaction's mechanism. Remember that the C-H and C-LG bonds must be antiperiplanar (180°) in order for the E2 mechanism to work.
Drawing out Newman projections is a great way to imagine this. What do you notice when we do that?
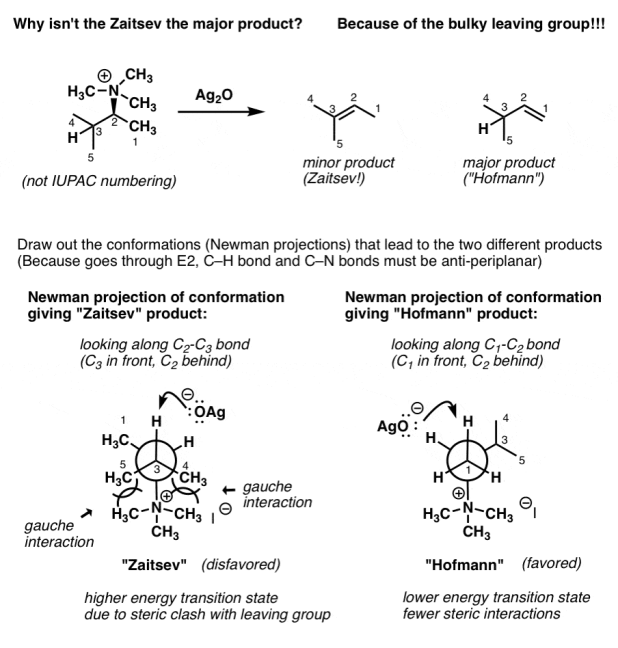
The steric hindrance of the leaving group isn't a consideration to consider in most elimination reactions. Despite the fact that leaving groups such as I and Br have a broad Van Der Waals radius, their bonds to carbon are long, and they are single atoms, so they do not interact with adjacent groups.
Contrast that to the NR3 group, which is like a big-ass ceiling fan spinning around its three alkyl groups – and each of the alkyl groups themselves is like a mini-ceiling fan spinning around three hydrogen atoms. It occupies a significant amount of space!
Because of the extremely bulky N(CH3)3 leaving group, the conformation that leads to the “Zaitsev” product has a lot more steric hindrance (two gauche interactions!) than the conformation that leads to the “Hofmann” product!
These additional steric interactions are sufficient to favour the Hofmann transition state over the Zaitsev transition state, resulting in the Hofmann product becoming the major product.
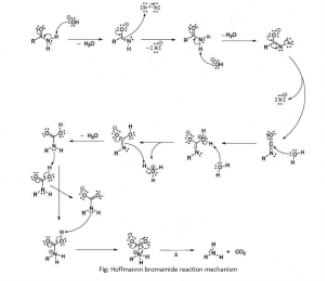
1.6.2 What Is Hoffmann bromamide Reaction?
As bromine is added to an amide in an aqueous or ethanolic sodium hydroxide solution, the amide degrades, resulting in the formation of primary amine. Hoffmann bromamide degradation reaction is a reaction that involves the degradation of an amide. The number of carbon atoms in the primary amine thus formed is one less than the number of carbon atoms in the amide.
RCONH2 + Br2 + 4NaOH R-NH2 + Na2CO3 + 2NaBr + 2H2O R-NH2 + Na2CO3 + 2NaBr + 2H2O
1.6.3 What Is the General Mechanism of Hoffmann Bromamide Reaction?
The following steps make up the general mechanism of the Hoffmann bromamide reaction:
The amide is attacked by a strong base (usually an alkali), which causes deprotonation and the formation of an anion, which then reacts with bromine to form N-bromoamide. This is an example of a -substitution reaction.
The bromoamide molecule is deprotonated to produce a bromoamide anion.
The R group attached to the carbonyl carbon migrates to nitrogen as the formed bromoamide anion undergoes rearrangement. Isocyanate is formed when the bromide ion leaves the compound at the same time.
To make carbamic acid, water molecules are added to the isocyanate. Nucleophilic addition is exemplified by this reaction.
Finally, the carbamic acid releases carbon dioxide, which causes primary amine to form.
1.7.1 What is Hofmann Elimination?
Hofmann elimination is the method of producing tertiary amines and alkenes from quaternary ammonium, as previously stated. Exhaustive methylation is another name for this procedure. When it comes to asymmetrical amines, the Hofmann rule states that the main alkene product is the least substituted and least stable. The Hofmann elimination method is named after its discoverer, August Wilhelm Von Hofmann, a German chemist. The Hofmann removal is depicted in the diagram below.
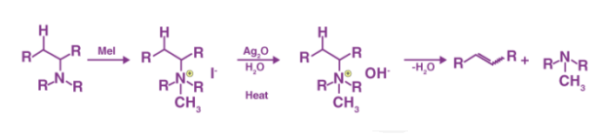
It's worth noting that methyl iodide is overused because it lacks beta hydrogens and hence cannot compete in the elimination reaction. The alkene isomer with the least substituted double bond is formed as the major product if the given alkyl group contains two separate sets of beta hydrogens that are available for the elimination process. The synthesis of trans-cyclooctene, shown below, is an interesting example of the Hofmann elimination process or exhaustive methylation process.
It's worth noting that the neutral NR3 group outperforms the anionic NH2– and NR2– groups as a leaving group. Steric effects caused by large leaving groups also have an impact on the reaction's outcome.
1.7.2 Hofmann Elimination Mechanism
The exhaustive methylation mechanism, also known as the Hofmann elimination mechanism, begins with the amine attacking the methyl iodide salt with a beta-hydrogen to form the ammonium iodide salt. Silver iodide is formed when the iodide reacts with the silver oxide. Since this silver iodide is insoluble, it precipitates out of solution. The water is deprotonated by a silver oxide ion, which forms a hydroxide ion. This mixture is currently being heated. The removal reaction is aided by the sun, and the hydroxide now picks up the beta-hydrogen from the ammonium ion. It also produces an amine, which is used to make the final alkene product. The following steps can be used to demonstrate the mechanism:
Step 1: As shown below, the ammonium iodide salt is formed by treating an amine with a beta-hydrogen or quaternary ammonium with excess methyl iodide.
Step 2 The iodide ion is substituted for the hydroxide ion by a reaction between the iodide and silver oxide, followed by deprotonation of water by the silver oxide ion. This stage is depicted in the diagram below.
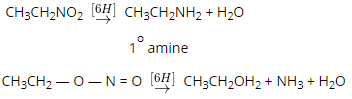
Step 3
The mixture is heated to facilitate and elimination reaction and form the required product.
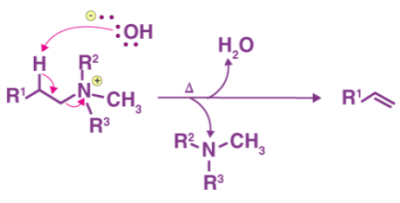
Thus, the required olefin product is achieved. The tertiary amine product is also formed. The Hofmann elimination process can be categorized as an elimination reaction as well as an olefination reaction.
1.8.1 What is Hinsberg Reagent?
Benzene sulfonyl chloride is also used as Hinsberg reagent. This name comes from its use in the Hinsberg test, which is used to detect and distinguish primary, secondary, and tertiary amines in a sample.
An organosulfur compound is what this reagent is. C6H5SO2Cl is the chemical formula for this substance. Hinsberg reagent has the appearance of a colourless oil that is viscous in nature and soluble in organic solvents.
This Reagent reacts with compounds that contain reactive O-H and N-H bonds. It is used to make sulfonamides and sulfonamide esters (by reacting with amines) (via reaction with alcohol).
1.8.2 Preparation of Hinsberg Reagent
The needed reagent is obtained by chlorinating benzene sulfonic acid or benzene sulfonic acid salts with phosphorus oxychloride (POCl3).
Reacting benzene with chloro sulfuric acid is another way to make the appropriate Hinsberg Reagent (chemical formula HSO3Cl). Both of these methods for preparing the necessary reagent are shown in the diagram below.
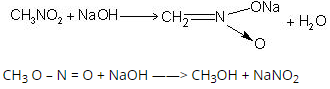
1.8.3 Hinsberg Test
The Hinsberg test is a chemical reaction that determines whether an amine is primary, secondary, or tertiary. Oscar Heinrich Daniel Hinsberg, a German chemist, was the first to describe this reaction in 1890.
The amines behave as nucleophiles in the Hinsberg Test, attacking the electrophile (sulfonyl chloride). This causes the chloride to be displaced, resulting in the formation of sulfonamides. As primary and secondary amines combine to form sulfonamides, the resulting sulfonamide is insoluble and precipitates as a solid from the solution.
1.8.4 Hinsberg Reaction Pathways
As benzene sulfonyl chloride reacts with primary amines, a sulfonamide compound is formed that is alkali soluble. The following diagram depicts this reaction.
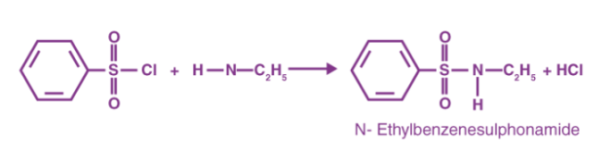
As benzene sulfonyl chloride reacts with secondary amines, a sulfonamide compound is formed that is not alkali soluble. Below is an example of this form of reaction.
Between a tertiary amine and the benzene sulfonyl chloride reagent, no such reaction occurs. Sulfonyl chloride hydrolysis is aided by tertiary amines. This reaction produces salts that are water soluble.
As a result, the Hinsberg reagent reacts differently with principal, secondary, and tertiary amines. These variations can be seen in the sulfonamide product's alkali solubility.
1.8.5 Reaction of Amines with Nitrous Acid
Nitrous acid (HNO2 or HONO) reacts with aliphatic amines in a fashion that provides a useful test for distinguishing primary, secondary and tertiary amines.
1°-Amines + HONO (cold acidic solution)  Nitrogen Gas Evolution from a Clear Solution
Nitrogen Gas Evolution from a Clear Solution
2°-Amines + HONO (cold acidic solution)  An Insoluble Oil (N-Nitrosamine)
An Insoluble Oil (N-Nitrosamine)
3°-Amines + HONO (cold acidic solution)  A Clear Solution (Ammonium Salt Formation)
A Clear Solution (Ammonium Salt Formation)
Nitrous acid is a Brønsted acid of moderate strength (pKa = 3.3). Because it is unstable, it is prepared immediately before use in the following manner:

Under the acidic conditions of this reaction, all amines undergo reversible salt formation:

This happens with 3º-amines, and the salts are usually soluble in water. The reactions of nitrous acid with 1°- and 2°- aliphatic amines may be explained by considering their behavior with the nitrosonium cation, NO (+), an electrophilic species present in acidic nitrous acid solutions.
Primary Amines

Secondary Amines

The action of 1o, 2o, and 3o-aliphatic amines is instructive in terms of understanding their chemistry, but it is of little use as a synthetic method. When branched primary alkyl groups are involved, SN1 product mixtures from 1o-amines are difficult to regulate, and rearrangement is normal. N-nitrosamines, which are derived from 2o-amines, are carcinogenic and are seldom used as intermediates in subsequent reactions.
Aryl Amines
The diazonium species produced by nitrous acid reactions of 1o-aryl amines are relatively stable and can be used as intermediates in a variety of aromatic substitution reactions. Resonance contributors can be used to classify dizonium cations, as shown in the bracketed formulas below. Since it has more bonding, the left-hand contributor is more powerful. Since the C-N bond is stronger and aryl carbocations are more stable than aliphatic 1o-amines, nitrogen loss is slower.

These diazonium ions are stable enough in aqueous solutions at 0o to 10oC to be used as intermediates in a number of nucleophilic substitution reactions. When water is the only nucleophile available for a reaction, for example, phenols are formed in high yield.
2º-Aryl Amines:
When 2o-aryl amines react with nitrous acid, they form N-nitrosamine derivatives and behave similarly to their aliphatic counterparts.
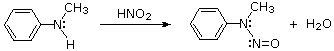
3º-Aryl Amines:
3o-Aryl amines can undergo aromatic ring nitrosation at sites ortho or para to the amine substituent, depending on ring substitution. Since the nitrosonium cation is not electrophilic enough to react with benzene or even toluene, strongly activated aromatic rings like amines and phenols can be substituted. Of course, as shown in the previous example, the rate of reaction of NO (+) directly at nitrogen is faster than that of ring substitution. The activating character of the amine nitrogen is greatly diminished once nitrosated, and ring nitrosation does not occur in N-nitrosoaniline derivatives, or indeed any amide derivative.
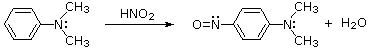
1.8.6 Reactions of Aryl Diazonium Salts
Substitution with Loss of Nitrogen
The salts of aryl diazonium are essential intermediates. They're made in a cold (between 0 and 10 oC) aqueous solution and react with nucleophiles to release nitrogen. The following diagram depicts some of the most widely used substitution reactions. These reactions are energetically preferred because the leaving group (N2) is thermodynamically very stable. Sandmeyer reactions are those substitution reactions that are catalysed by cuprous salts. Treatment with BF4(–) causes fluoride substitution, which is known as the Schiemann reaction. It is possible to isolate stable diazonium tetrafluoroborate salts, which lose nitrogen when heated to produce an arylfluoride product. The reductive elimination of an amino (or nitro) group is achieved in the top reaction with hypophosphorus acid, H3PO2. This reduction, unlike nucleophilic substitution reactions, is most likely the product of a radical process.
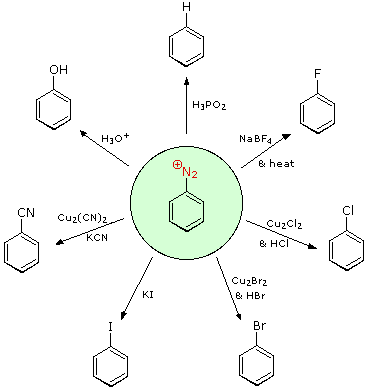
These aryl diazonium substitution reactions vastly increase the number of polysubstituted benzene derivatives that can be synthesised. Think of the following possibilities:
I The corresponding nitro compound is the most common precursor to an aryl amine. An aromatic ring is deactivated by a nitro substituent, which guides electrophilic substitution to meta locations.
(ii) There are many methods for reducing a nitro group to an amine. An aromatic ring is strongly activated by the amine substituent, which guides electrophilic substitution to ortho and para positions.
(iii) By forming an amide derivative (reversible), the activating character of an amine substituent may be reduced, or even modified to deactivating and meta-directing by forming a quaternary-ammonium salt (irreversible).
(iv) By converting an aryl amine to a diazonium ion intermediate, a number of different groups (including hydrogen) can be substituted, which can then be used in subsequent reactions.

Bonding to Nitrogen
The positive charge is delocalized over the two nitrogen atoms, according to a resonance description of diazonium ions. Nucleophiles cannot bind to the inner nitrogen, but negative nucleophiles will bond (or couple) to the terminal nitrogen to produce neutral azo compounds. This coupling to the terminal nitrogen should be relatively quick and reversible, as seen in the following equation. E / Z stereoisomers of the azo products can exist. In fact, the E-isomer is found to predominate at equilibrium.

Reversal to the diazonium ion and slow nucleophilic substitution at carbon (with irreversible nitrogen loss) would be the ultimate course of reaction unless these azo products are trapped or stabilised in some way, as stated in the previous section. As phenyldiazonium bisufate is quickly added to a cold sodium hydroxide solution, a relatively stable solution of sodium phenyldiazoate (the conjugate base of the initially formed diazoic acid) results. Lowering the pH of this solution produces phenyldiazoic acid (pKa ca. 7), which dissociates back to the diazonium ion and then undergoes substitution to produce phenol.
C6H5N2(+) HSO4(–) + NaOH (cold solution) |  | C6H5N2–OH + NaOH (cold) |  | C6H5N2–O(–) Na(+) |
phenyldiazonium bisulfate |
| phenyldiazoic acid |
| sodium phenyldiazoate |
Aryl diazonium salts may be reduced to the corresponding hydrazines by mild reducing agents such as sodium bisulfite, stannous chloride or zinc dust. The bisulfite reduction may proceed by an initial sulfur-nitrogen coupling, as shown in the following equation.
Ar-N2(+) X (–) NaHSO3
Ar-N=N-SO3H NaHSO3
Ar-NH-NH-SO3H H2O
Ar-NH-NH2 + H2SO4
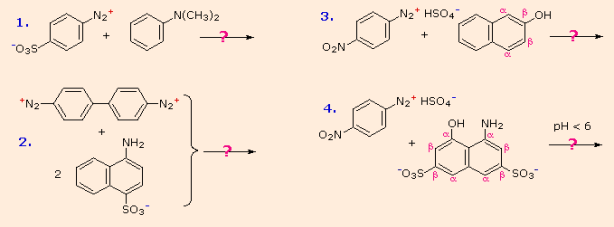
Electrophilic aromatic substitution of activated benzene derivatives by diazonium electrophiles is the most common application of diazo coupling reactions. These reactions produce highly coloured aromatic azo compounds, which are used as synthetic dyestuffs and are known as azo dyes. The colour of azobenzene (Y=Z=H) is light orange; however, depending on the nature of the aromatic rings and the substituents they bear, the colour of other azo compounds can vary from red to deep blue. Although there are cis/trans isomer pairs of azo compounds, the majority of well-characterized and stable compounds are trans.

The following are some examples of azo coupling reactions. A few basic rules will help predict how certain reactions will unfold:
An amino group is a better activating substituent than a hydroxyl group at acid pH (6). (i.e., a phenol). Due to increased phenoxide base concentration, phenolic functions are stronger activators at alkaline pH (> 7.5).
If that position is open, coupling to an activated benzene ring occurs preferentially para to the activating group. If this does not happen, ortho-coupling will occur.
(iii) Naphthalene undergoes electrophilic substitution at alpha sites more quickly than at beta sites, but ortho-coupling is favoured. Examples of / notation in naphthalene’s can be found in the diagram.
1.8.7 Substitution and Elimination Reactions of Amines
In nucleophilic substitution or base-catalyzed elimination reactions, amino functions are rarely used as leaving groups. Indeed, they are even less powerful than hydroxyl and alkoxyl groups in this regard. Modifying the leaving group (OH (–) or OR (–) to increase its stability as an anion was a useful technique for increasing the reactivity of the oxygen function in alcohols and ethers (or equivalent). The power of the corresponding conjugate acids can be used to measure this stability.
As previously mentioned, 1o and 2o-amines are much weaker acids than alcohols, so it's not shocking that forcing the nitrogen mechanism to act as a nucleophilic leaving group is difficult. Heating an amine with HBr or HI, for example, does not usually result in the formation of the corresponding alkyl halide, as it does with alcohols and ethers. The acidity of the putative ammonium leaving group is at least ten powers of ten lower than that of an equivalent oxonium species in this sense. Due to the extreme stability of this leaving group, nitrogen loss from diazonium intermediates is a significant exception in this relation (the conjugate acid of N2 would be an extraordinarily strong acid).
The tetraalkyl (4o-) ammonium salts are one class of amine derivatives that has proven useful in SN2 and E2 reactions. The majority of applications involving this class of compounds are eliminations, although there have been a few reports of SN2 substitution.
acetone & heat
C6H5–N(CH3)3(+) Br (–) + R-S (–) Na (+)  R-S-CH3 + C6H5–N(CH3)2 + NaBr
R-S-CH3 + C6H5–N(CH3)2 + NaBr
heat
(CH3)4N (+) OH (–) CH3–OH + (CH3)3N
CH3–OH + (CH3)3N
Hofmann Elimination
Hofmann eliminations are 4o-ammonium salt reduction reactions. Since the counter anion in most 4o-ammonium salts is halide, it is frequently replaced by the more simple hydroxide ion via silver hydroxide reaction (or silver oxide). To impact the E2-like removal of a 3o-amine, the resulting hydroxide salt must be heated (100 - 200 oC). A typical Hofmann elimination is shown in Example #1 below. As previously mentioned, when looking at alkyl halide elimination reactions, one of the alkyl substituents on nitrogen must have one or more beta-hydrogens for an elimination to occur.
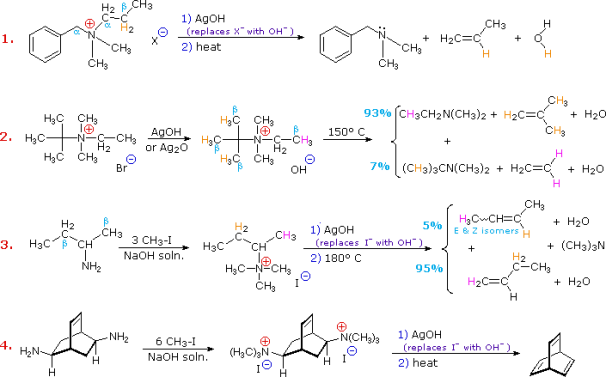
Two of the alkyl substituents on nitrogen in Example #2 have beta-hydrogens, all of which are on methyl groups (colored orange & magenta). The alkene with the more strongly substituted double bond is the main product of the removal, reflecting not only the 3:1 numerical advantage of certain beta-hydrogens, but also the greater stability of the double bond.
Example #3 highlights two main aspects of the Hofmann elimination:
First, by extensive alkylation, typically with methyl iodide, simple amines are easily converted to the necessary 4o-ammonium salts (methyl has no beta-hydrogens and cannot compete in the elimination reaction). In example #4, exhaustive methylation is demonstrated once more.
Second, when an alkyl group has two sets of beta-hydrogens available for removal (coloured orange and magenta here), the alkene isomer with the less substituted double bond is always the main product.
The Hofmann Rule refers to the tendency of Hofmann eliminations to produce the less-substituted double bond isomer, which contrasts sharply with the Zaitsev Rule for dehydrohalogenations and dehydrations. The Hofmann Rule does not apply when other triggering groups, such as phenyl or carbonyl, are present. Thus, if 2-amino-1-phenylpropane is handled in the manner of example #3, the result consists largely of 1-phenylpropene (E & Z-isomers) (E & Z-isomers).
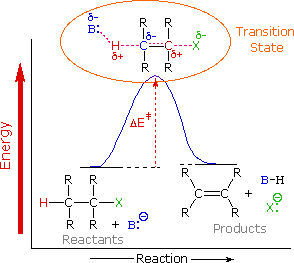
It's important to re-examine the existence of the E2 transition state, which was first identified for dehydrohalogenation, to understand why base-induced removal of 4o-ammonium salts behaves differently than that of alkyl halides. On the right, the energy diagram for a single-step bimolecular E2 process is repeated. E2 reactions have a less well-defined transition state than SN2 reactions. More bonds are breaking and forming, with the probability of a continuum of states where the degree of C–H and C–X bond breaking and C=C bond formation varies. The transition state resembles that of an E1 reaction if the bond to the leaving group (X) is significantly broken relative to the other bond changes (initial ionisation followed by a fast second step). On the other hand, if the beta-hydrogens' acidity is increased, substantial C–H bond breaking may occur before the other bonds are affected. For most simple alkyl halides, it was necessary to imagine a balanced transition state in which all bonds changed at the same time. The Zaitsev Rule was compatible with such a model.
When the leaving group X has a positive charge, as it does in the 4o-ammonium compounds discussed here, the inductive effect of the charge increases the acidity of both the alpha and beta-hydrogens. Furthermore, since the 4o-ammonium substituent is far larger than a halide or hydroxyl group, it can disrupt the conformations that substituted beta-carbons may take. It appears that a combination of these factors favours base attack on the beta-hydrogens with the least substituted (least hindered and acidic) group of substituents. Many Hofmann eliminations have the preferred anti orientation of the leaving group and beta-hydrogen, as seen in dehydrohalogenation; however, syn-elimination is also normal, likely due to the attraction of opposite charges orienting the hydroxide base near the 4o-ammonium leaving group.
In the diagram below, three more examples of the Hofmann elimination are given. In two ways, Example #1 is intriguing. For starters, it produces a 4o-ammonium halide salt in a different way than exhaustive methylation. Second, prior to elimination, this salt is not converted to its hydroxide analogue. The volatile hydrocarbon substance is isolated by distillation after a concentrated aqueous solution of the halide salt is lowered into a refluxing sodium hydroxide solution.
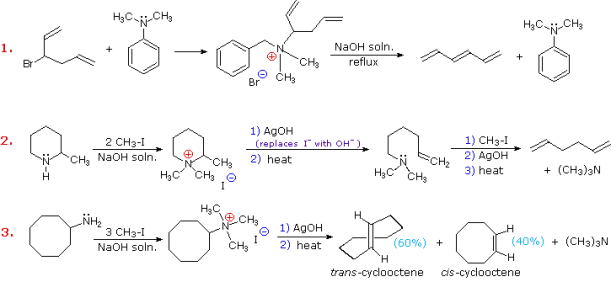
Example #2 exemplifies a key feature of the Hofmann elimination. If the nitrogen atom is part of a ring, this removal technique does not extract the nitrogen as a separate 3o-amine product in a single application. A second Hofmann elimination is needed to separate the nitrogen feature from the molecule. Indeed, if the nitrogen atom was part of two rings (fused or spiro), it would take three Hofmann eliminations to separate the nitrogen from the remaining molecular structure.
The less stable trans-cyclooctene is the main product in Example #3, which is followed by the cis-isomer. Since the cis-cycloalkene will be produced by an anti-E2-transition state, the trans-isomer must be produced by a syn-elimination. A syn-elimination may also generate the cis-cyclooctene released in this reaction. The structure of cyclooctane is conformationally complex. There are a number of puckered conformations that escape angle strain, and one of the most stable is shown on the right. Many of these conformers have several eclipsed bonds, and transannular hydrogen crowding is inevitable. Since the trimethylammonium substituent is wide (roughly the same size as tert-butyl), it will most likely take on an equatorial orientation to prevent steric crowding. An axial-like orientation of this bulky group is likely needed for an anti-E2 transition state, making this an unfavourable direction.
1.8.8 Oxidation States of Nitrogen:
When comparing the chemistry of amines with that of alcohols and ethers, we find a number of similar compounds in which nitrogen takes on higher oxidation states than oxygen, which has a reduced number of oxidation states. Bear in mind that the oxidation states of elemental oxygen (O2) and nitrogen (N2) are both zero in this sense.
The most common covalently bonded oxygen state is -2. Air, alcohols, ethers, and carbonyl compounds are examples of this. The peroxides R–O–O–R, where R=hydrogen, alkyl, aryl, or acyl, have the only higher oxidation state (-1). These compounds are commonly used as free radical initiators due to the low covalent bond energy of the peroxide bond (ca.35 kcal/mole), and their reactivity can be dangerously explosive (e.g., triacetone triperoxide used by terrorist bombers).
Nitrogen compounds, on the other hand, cover a wide range of nitrogen oxidation states, from -3 (as in ammonia and amines) to +5 (as in nitric acid). The table below lists some of the known organic nitrogen compounds with various oxidation states of that element. Some of these chemical groups have already been discussed; others will be discussed later.
Oxidation State | _3 | _2 | _1 | 0 | +1 | +3 |
Formulas | R3N (amines) | R2N–NR2 (hydrazines) | RN=NR (azo cpd.) | N2 (nitrogen) | R–N=O (nitroso) | R-NO2 (nitro) |
Amine Oxides
Amine oxides are prepared by oxidizing 3º-amines or pyridines with hydrogen peroxide or peracids (e.g., ZOOH, where Z=H or acyl).
R3N: + ZOOH R3N (+)–O (–) + ZOH
R3N (+)–O (–) + ZOH
In comparison to the parent amine, amine oxides are relatively weak bases, with a pKa of about 4.5. The polar coordinate covalent N–O function has oxygen as a strong hydrogen bond acceptor. The amine oxide formed when one of the alkyl substituents has a long chain, such as C12H25, is an amphoteric surfactant that is used in shampoos and other mild cleaning agents.
When 3o-amine oxides are heated to temperatures of 150 to 200 oC, an elimination reaction occurs that is complementary to the Hofmann elimination. The Cope Elimination is the name for this reaction. Dropwise addition of an amine oxide solution to a heated tube packed with small glass beads is a popular method. The volatile alkene products are carried to a chilled receiver by a current of nitrogen gas flowing through the column. A hydroxyl amine is the nitrogen-containing product. Unlike the Hofmann elimination, this reaction is catalysed by a concerted cyclic reorganisation, as depicted in the diagram below. The beta-hydrogen and amine oxide moieties must have a syn-relationship for such a system to work.
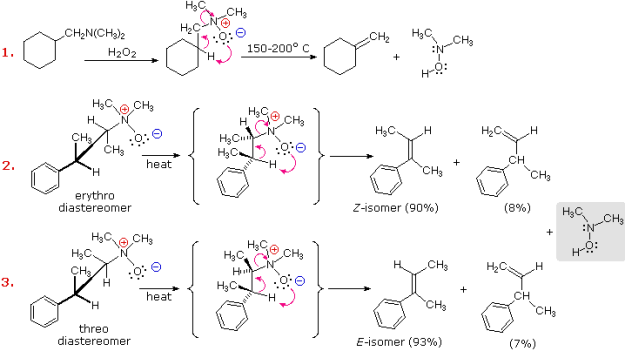
Cope removal of diastereomeric amine oxides, as seen in examples #2 and 3 above, demonstrates the beta-hydrogen and amine oxide groups' syn-relationship. These examples also show that the more stable double bond has a high regioselectivity.
Nitroxide Radicals
Peroxides (ZOOH) oxidise 2o-amines lacking -hydrogens to nitroxide radicals with remarkable stability. It should be remembered that resonance delocalization of the unpaired electron leads to a polar N–O bond in the example shown at the top of the following diagram. The R=H compound, also known as TEMPO, is a red solid that is relatively stable. There have been several other nitroxides prepared, three of which are depicted in the lower right corner. The nitroxide decomposes to mixtures of amine oxides and nitrones if one or more hydrogens are present on an adjacent carbon, as seen at the lower left. Halogens oxidise nitroxides to unstable oxammonium cations.
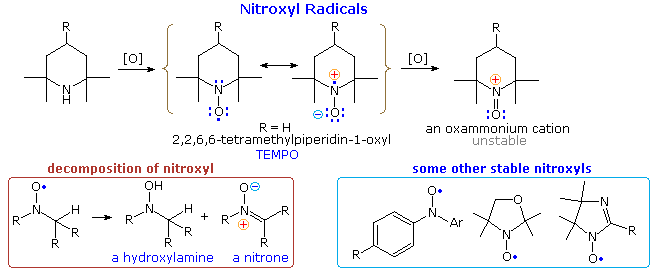
A technique known as electron paramagnetic resonance can be used to investigate the spin of the nitroxyl unpaired electron (epr or esr). Experiments of this kind have shown that the epr spectra are responsive to the radical's substituents as well as its immediate surroundings. This has resulted in the creation of a spin labelling technique for studying the conformational structures of macromolecules such as proteins. As a result, site-directed spin labelling (SDSL) has emerged as a useful technique for mapping secondary structure at the backbone fold level in a wide range of proteins, including those that aren't amenable to structural characterization using traditional structural techniques like nuclear magnetic resonance and X-ray crystallography.
References:
1. Graham Solomons T. W., Fryhle, Craig B., Snyder Scott A, Organic Chemistry, Wiley
Student Ed, 11th Edition (2013)
2. Jonathan Clayden, Nick Greeves, Stuart Warren, Organic Chemistry, 2nd Edition, Oxford
Publisher, 2014.
3. Dhawan, S.N., Pradeep’s Organic Chemistry, (Vol. I and II), Pradeep Publications




