Unit - 3
Heterocyclic Compounds
3.1.1 Definition:
Heterocyclic compounds are the largest and most diverse group of organic compounds. After all, any carbocyclic compound, regardless of structure or functionality, can theoretically be transformed into a series of heterocyclic analogues by substituting a different element for one or more of the ring carbon atoms. Even if we limit ourselves to oxygen, nitrogen, and sulphur (the three most common heterocyclic elements), the possibilities for such a substitution are endless.
3.1.2 Nomenclature:
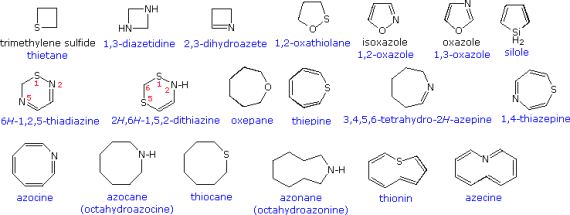
Developing a formal nomenclature scheme for heterocyclic compounds was a difficult task that has yet to be completed. Many heterocycles, especially amines, were discovered early on and given incomprehensible names that are still used today. The following chart shows several monocyclic compounds of this type, with the common (trivial) name in bold and a systematic name based on the Hantzsch-Widman method beneath it in blue. The guidelines for using this framework will be given at a later time. For the most part, studying these traditional names will suffice as a nomenclature foundation.

An elemental prefix for the heteroatom is preceded by the relevant carbocyclic name in a simple but restricted nomenclature scheme. In the table below, a short list of some common prefixes is listed in priority order from right to left. Ethylene oxide = oxacyclopropane, furan = oxacyclopenta-2,4-diene, pyridine = azabenzene, and morpholine = 1-oxa-4-azacyclohexane are examples of this nomenclature.
Element | oxygen | sulfur | selenium | nitrogen | phosphorous | silicon | Boron |
Valence | II | II | II | III | III | IV | III |
Prefix | Oxa | Thia | Selena | Aza | Phospha | Sila | Bora |
The Hantzsch-Widman scheme is a more systematic way of naming heterocyclic compounds that isn't reliant on carbocyclic names. It starts with the same hetero atom prefix as before (minus the final "a"), then adds a suffix that specifies ring size and saturation. Each suffix has a ring size root (blue) and an ending that designates the degree of unsaturation in the ring, as shown in the table below. It's important to note that the saturated suffix only applies to fully saturated ring structures, while the unsaturated suffix only applies to rings with the most non-cumulated double bonds. A prefix such as "dihydro" or "tetrahydro" is required for systems with a lower degree of unsaturation.
Ring Size | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
Suffix |
|
|
|
|
|
|
|
|
Several exceptions and revisions have been added to accommodate problems with previous use, despite the Hantzsch-Widman system's general systematic structure. Here are a few examples:
• The "e" at the end of the suffix is optional, but recommended.
• Saturated 3, 4, and 5-membered nitrogen heterocycles should be suffixed with "iridine," "etidine," and "olidine," respectively.
• The conventional "irine" suffix can be used on unsaturated nitrogen 3-membered heterocycles.
• The former use of "etine" and "oline" as suffixes for identical sized nitrogen heterocycles prevents their use as a suffix for 4 and 5-membered unsaturated heterocycles.
• Since oxine, azine, and silane have been used for other compounds or purposes, they cannot be used for pyran, pyridine, or silacyclohexane.
In both the preceding and the following diagrams, examples of these nomenclature rules are written in blue. To prevent ambiguity, the position of a saturated atom in a maximally unsaturated ring can be marked by a "#H " prefix, as in pyran and pyrrole above and many examples below. When numbering a ring of more than one heteroatom, the highest priority atom is numbered first, followed by the next highest priority atom, and so on.
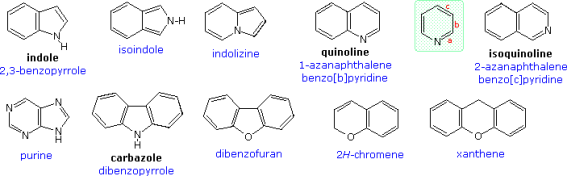
Many of the compounds in the preceding examples were monocyclic. One or more heterocyclic rings are commonly found in polycyclic compounds. A few of these are depicted in the illustration below. Popular names are in black, and systematic names are in blue, as before. The two quinolines show a different side of heterocyclic nomenclature. As shown by the pyridine ring in the green shaded box, the position of a fused ring can be indicated by a lowercase letter that designates the edge of the heterocyclic ring involved in the fusion.
Many naturally occurring compounds contain heterocyclic rings. They are responsible for the main structures of mono and polysaccharides, as well as the four DNA bases that make up the genetic code. Other examples of heterocyclic natural products will be shown if you click on the diagram above.
3.1.3 Preparation and Reactions:
Three-Membered Rings
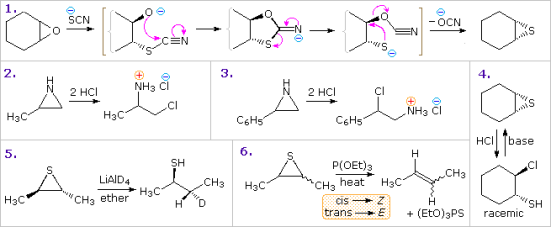
The most frequently found three-membered heterocycles are oxiranes (epoxides). Epoxides are easily made by reacting alkenes with peracids, with strong stereospecificity in most cases. Epoxides are more reactive than unstrained ethers due to the high angle strain of the three-membered ring. The most general reaction class is addition reactions that include the electrophilic or nucleophilic opening of the ring. One such transformation is seen in Example 1 in the diagram below, which is interesting due to the subsequent conversion of the addition intermediate into the corresponding thiirane. The intermediate relaxes to the diequatorial conformer before cyclizing to a 1,3-oxathiolane intermediate, which is stereo electronically guided in a trans-diaxial fashion.
Related addition reactions to thiiranes and aziridines can be seen in other examples. The acid-catalyzed additions in examples 2 and 3 demonstrate how substituents affect addition regioselectivity. The SN2 character of nucleophile (chloride anion) attack on protonated aziridine is reflected in Example 2. (The less substituted carbon is the site of addition). In Example 3, the phenyl substituent stabilises the forming carbocation to the point where SN1 selectivity is achieved. The stereospecific reduction of thiiranes to alkenes by reaction with phosphite esters (example 6) is thought to occur due to an initial bonding of phosphorous to sulphur.
Four more examples of three-membered heterocycle reactivity or intermediacy will be shown if you click on the diagram above. Thermal reactions in which both the heteroatom and the strained ring play a role are examples 7 and 8. In the solvolysis of optically active 2-bromopropanoic acid (example 9), the -lactone intermediate accounts for both the 1st-order kinetics of this reaction and the preservation of configuration in the product. It's worth noting that two configuration inversions at C-2 result in overall retention. Many examples of intramolecular interactions have been documented, including Example 10.
The intramolecular ring-opening reactions of disubstituted epoxides with a pendant -hydroxy substituent have shown an interesting regioselectivity. Acid and base-catalyzed reactions usually proceed by 5-exo-substitution (reaction 1), resulting in a tetrahydrofuran product, as shown below. If the oxirane has an unsaturated substituent (vinyl or phenyl), the acid-catalyzed opening takes place in a 6-endo pattern at the allylic (or benzylic) carbon (reaction 2). The substituent's -electron system aids positive charge production at the adjacent oxirane carbon, directing nucleophilic attack to that site.

Four-Membered Rings
● Preparation
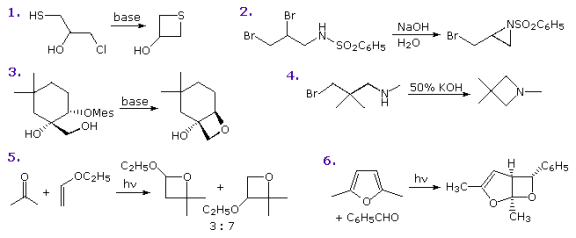
The following diagram depicts some methods for preparing four-membered heterocyclic compounds. Treatment of a 3-halo alcohol, thiol, or amine with base is usually reliable, but yields are often mediocre. Side reactions such as dimerization and elimination are normal, and other functions can compete in the reaction. In Example 1, cyclization to an oxirane competes with thietane formation, but sulfur's greater nucleophilicity wins out, especially if a weak base is used. Both aziridine and azetidine can be formed in Example 2, but only the former is observed. This is an excellent illustration of the kinetic benefit of three-membered ring formation. In the absence of competition, Example 4 shows that this approach to azetidine formation works well. The gem-dimethyl substitution, also known as the Thorpe-Ingold effect, is thought to favour coiled chain conformations, which is why this product has such a high yield. The substrate in Example 3 has a relatively rigid structure that favours oxetane formation and prevents oxirane cyclization. Finally, the Paterno-Buchi photocyclizations in examples 5 and 6 are well-suited to the formation of oxetane.
● Reactions
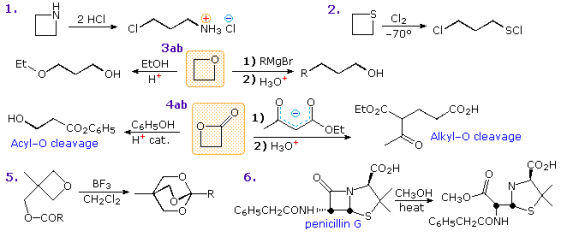
The effect of ring strain can also be seen in reactions involving four-membered heterocycles. In the diagram below, some examples are given. As seen in examples 1, 2 and 3a, acid catalysis is a typical feature of many ring-opening reactions. In the thietane reaction (2), the sulphur is electrophilically chlorinated to form a chlorosulfonium intermediate, which is then substituted with a ring-opening chloride ion. As shown in reaction 3b, strong nucleophiles can also open the strained ether. Acid-catalyzed acyl exchange, as in 4a, or alkyl-O rupture by nucleophiles, as in 4b, are also possible cleavage reactions for -lactones. A fascinating case of intramolecular rearrangement to an ortho-ester is Example 5. Finally, the enhanced acylating reactivity of this fused ring mechanism is demonstrated by the -lactam cleavage of penicillin G (reaction 6). Because of p-resonance stabilisation, most amides are highly unreactive acylation reagents. In penicillins, such electron pair delocalization is reduced, resulting in a pyramidal nitrogen structure and a more reactive carbonyl feature against nucleophiles.
Five-Membered Rings
● Preparation
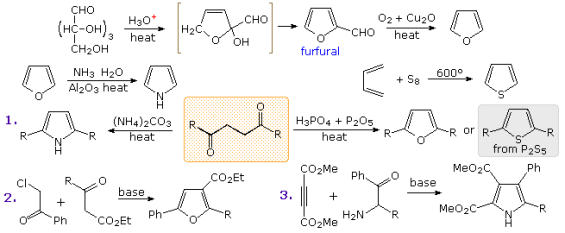
As shown in the uppermost equation below, commercial preparation of furan begins with the aldehyde furfural, which is generated from pentose-containing raw materials such as corncobs. The second-row equations show pyrrole and thiophene preparations that are similar. The Paal-Knorr synthesis, shown in the third row, is a general method for making substituted furans, pyrroles, and thiophenes from 1,4-dicarbonyl compounds. There has been a slew of other procedures that lead to substituted heterocycles of this kind. Reactions 2 and 3 demonstrate two of these. Palladium-catalyzed hydrogenation reduces furan to tetrahydrofuran. Not only is this cyclic ether a useful solvent, but it can also be easily converted into 1,4-dihalobutanes or 4-haloalkylsulfonates, which can be used to make pyrrolidine and thiolane.
Dipolar cycloaddition reactions often result in five-membered heterocycles with more complexity.
Indole is probably the most important fused ring heterocycle in this class. By clicking on the above diagram three examples of indole synthesis will be displayed. The first proceeds by an electrophilic substitution of a nitrogen-activated benzene ring. The second presumably takes place by formation of a dianionic species in which the ArCH2(–) unit bonds to the deactivated carbonyl group. Finally, the Fischer indole synthesis is a remarkable sequence of tautomerism, sigmatropic rearrangement, nucleophilic addition, and elimination reactions occurring subsequent to phenylhydrazone formation. This interesting transformation involves the oxidation of two carbon atoms and the reduction of one carbon and both nitrogen atoms.
Reactions
The chemical reactivity of the saturated members of this class of heterocycles: tetrahydrofuran, thiolane and pyrrolidine, resemble that of acyclic ethers, sulfides, and 2º-amines, and will not be described here. 1,3-Dioxolanes and dithiolanes are cyclic acetals and thioacetals. These units are commonly used as protective groups for aldehydes and ketones, and may be hydrolyzed by the action of aqueous acid.
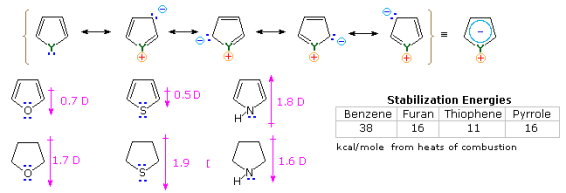
It is the "aromatic" unsaturated compounds, furan, thiophene and pyrrole that require our attention. In each case the heteroatom has at least one pair of non-bonding electrons that may combine with the four π-electrons of the double bonds to produce an annulene having an aromatic sextet of electrons. This is illustrated by the resonance description at the top of the following diagram. The heteroatom Y becomes sp2-hybridized and acquires a positive charge as its electron pair is delocalized around the ring. An easily observed consequence of this delocalization is a change in dipole moment compared with the analogous saturated heterocycles, which all have strong dipoles with the heteroatom at the negative end. As expected, the aromatic heterocycles have much smaller dipole moments, or in the case of pyrrole a large dipole in the opposite direction. An important characteristic of aromaticity is enhanced thermodynamic stability, and this is usually demonstrated by relative heats of hydrogenation or heats of combustion measurements. By this standard, the three aromatic heterocycles under examination are stabilized, but to a lesser degree than benzene.
Additional evidence for the aromatic character of pyrrole is found in its exceptionally weak basicity (pKa ca. 0) and strong acidity (pKa = 15) for a 2º-amine. The corresponding values for the saturated amine pyrrolidine are: basicity 11.2 and acidity 32.
Aromatic systems also have a pattern of reactivity with electrophilic reagents, which is particularly important to chemists. Simple cycloalkenes are more likely to react by addition, while aromatic compounds are more likely to react by substitution. These substitutions occur by an initial electrophile addition, followed by a proton loss from the "onium" intermediate to regenerate the aromatic ring, as seen with benzene and its derivatives. Electrophilic substitution occurs in all aromatic five-membered heterocycles, with the following reactivity order: pyrrole >> furan > thiophene > benzene. In the diagram below, some examples are given. The reaction conditions clearly indicate that furan has a higher reactivity than thiophene. All of these aromatic heterocycles react violently with chlorine and bromine, resulting in polyhalogenated materials that are often combined with polymers. The reaction of pyrrole with iodine (bottom left equation) and the formation of 2-acetylpyrrole by simply heating it with acetic anhydride demonstrate its exceptional reactivity (no catalyst).
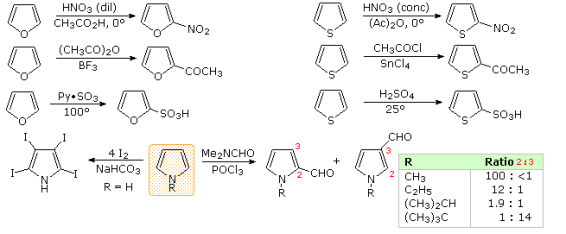
Especially for furan and thiophene, there is a strong preference for substitution at the ring's 2-position (). Since N-protonation removes the aromatic character of pyrrole, reactions involving it must be carefully evaluated. Indeed, prior to subsequent reactions, this 2o-amine is often N-substituted. N-acetylpyrrole is formed when pyrrole reacts with acetic anhydride or acetyl chloride and triethyl amine. As a result, as the bottom right equation shows, the regioselectivity of pyrrole substitution is variable.
The mechanism described below provides an explanation for the general -selectivity of these substitution reactions. Charge delocalization stabilises the intermediate formed by electrophile attack at C-2 to a greater extent than the intermediate formed by C-3 attack. We may deduce from the Hammond postulate that the activation energy for substitution at the first position is lower than the activation energy for substitution at the second position.
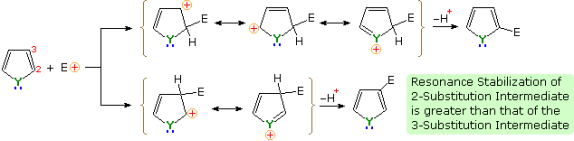
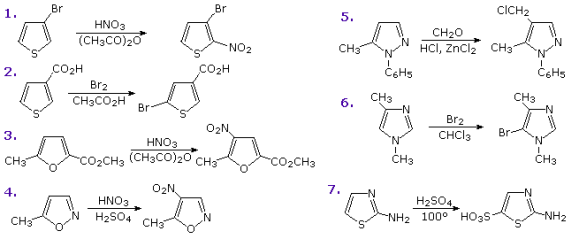
Functional substituents have the same effect on these heterocycles' substitution reactions as they do on benzene. It is not difficult to predict their directing effect on heterocyclic ring substitution once one recognises the ortho-para and meta-directing character of these substituents. Seven of these reactions are depicted in the diagram below. Reactions 1 and 2 are 3-substituted thiophenes with an electron donating substituent in the first and an electron withdrawing group in the second. In the second and fifth positions of the third reaction, there are two distinct forms of substituents. Finally, examples 4 through 7 show 1,2- and 1,3-oxazole, thiazole, and diazole reactions. The apparent sp3-hybridized nitrogen, whose electron pair is part of the aromatic electron sextet, has a basicity over a million times greater than the sp2-hybridized nitrogen in the diazoles.
The structural features of these heterocycles point to other potential reactions. Furan, for example, is an enol ether, while pyrrole is an enamine. Acid-catalyzed hydrolysis of such functions to carbonyl compounds, alcohols, or amines is known. We might expect Diels-Alder cycloaddition reactions with suitable dienophiles since these compounds are also heteroatom substituted dienes. By clicking on the diagram above, you can see all of these options. Furans can be hydrolyzed to 1,4-dicarbonyl compounds, as seen in the previous example, but pyrroles and thiophenes behave differently. In the centre, the second and third examples show typical reactions of furan and pyrrole with the heavy dienophile maleic anhydride. The former is involved in a cycloaddition reaction, while the pyrrole is purely electrophilic substituted at C-2. Thiophene has a difficult time reacting with this dienophile.
The bottom line of the new diagram shows how additional nitrogen units have a significant impact on the hydrolysis of a series of N-acetylazoles in water at 25°C and pH=7. The pyrrole compound on the left is basically unreactive, as one would assume for an amide, but adding nitrogens accelerates hydrolysis significantly. This effect has been applied to the acylation reagent 1,1'-carbonyldiimidazole (Staab's reagent) in practical applications.
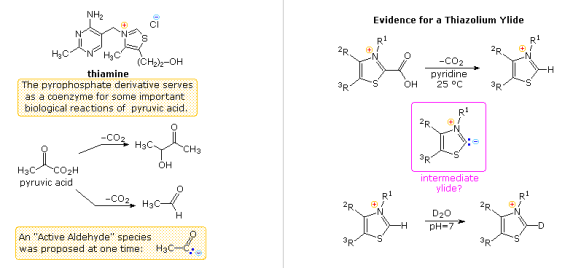
During research into the effects of thiamine, another aspect of heterocyclic chemistry was discovered (following diagram). Thiamine is a coenzyme for many biochemical processes, including the decarboxylation of pyruvic acid to acetaldehyde and acetoin, as its pyrophosphate derivative. An "active aldehyde" or acyl carbanion species was thought to be an intermediate in these reactions by early researchers. Many suggestions were made, some involving the aminopyrimidine moiety and others involving thiazole ring-opened hydrolysis derivatives, but none were satisfactory. R. Breslow (Columbia) discovered that the C-2 hydrogen of thiazolium salts was surprisingly acidic (pKa ca. 13), creating a relatively stable ylide conjugate base, which solved the puzzle. This explains the ease with which thiazolium-2-carboxylic acids can be decarboxylated and deuterium exchanged at C-2 in neutral heavy water.
In the same way that the cyanide ion catalyses the formation of benzoin from benzaldehyde, appropriate thiazolium salts catalyse the conversion of aldehydes to acyloins. A new window will appear when you click on the diagram, showing mechanisms for these two reactions. It's worth noting that an acyl anion equivalent is formed in both cases, which then adds to a carbonyl function in the expected way. While the use of thiazolium catalysts has proven to be useful for both aliphatic and aromatic aldehydes, the benzoin condensation is restricted to aromatic aldehydes. The reduction of esters to enediol intermediates by the action of metallic sodium in this approach to acyloins uses milder conditions.
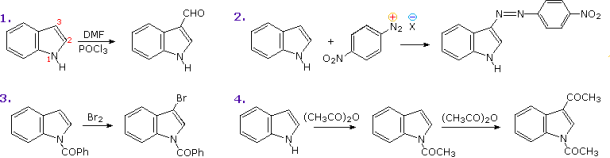
Indole is the most important condensed ring structure associated with these heterocycles. The following diagram depicts several electrophilic indole substitution reactions. If the indole nitrogen is substituted or not, the C-3 of the heterocyclic ring is the preferred attack site. The electrophile's bonding at that position allows the onium-intermediate to be stabilised by the nitrogen without disrupting the benzene aromaticity.
Six-Membered Rings
● Properties
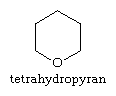
The reactivity of the saturated members of this class of heterocycles, such as tetrahydropyran, thiane, and piperidine, is similar to that of acyclic ethers, sulphides, and 2o-amines, and will not be discussed further. Dithianes and 1,3-dioxanes are cyclic acetals and thioacetals, respectively. These units are widely used as defensive groups for aldehydes and ketones, as well as synthetic intermediates, and are susceptible to aqueous acid hydrolysis. The interaction between the double bond and the heteroatom determines the reactivity of partially unsaturated compounds (e.g., 3,4-dihydro-2H-pyran is an enol ether).
The aromatic rings of completely unsaturated six-membered nitrogen heterocycles like pyridine, pyrazine, pyrimidine, and pyridazine are stable. As in the case of 2,4,6-triphenylpyrylium tetrafluoroborate, oxygen and sulphur analogues are still positively charged.
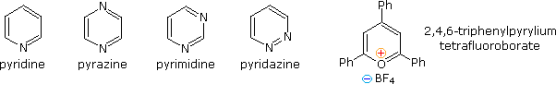
The aromatic stabilisation energy of pyridine is 21 kcal/mole, according to heat of combustion measurements. The charge separated structures not usually considered for benzene are included in the resonance definition drawn at the top of the following diagram. Given nitrogen's higher electronegativity (in comparison to carbon), such canonical forms can play a significant role. The fact that pyridine has a greater dipole moment than piperidine confirms this theory. Pyridine and its derivatives are poor bases, owing to the nitrogen's sp2 hybridization. It should be clear from the polar canonical types that electron donating substituents increase pyridine basicity, and that substituents on the 2 and 4-positions have a greater impact on basicity than an analogous 3-substituent. A few of these substituent effects are shown by the pKa values in the table. Picoline (methyl pyridines), lutidine (dimethyl pyridines), and collidine are popular names for methyl substituted derivatives (trimethyl pyridines). The effects of 2-substituents are complicated, with steric hindrance and electrostatic components. For acylation reactions in pyridine as a solvent, 4-dimethylaminopyridine is a useful catalyst. The sp3 hybridised nitrogen may appear to be the stronger base at first glance, but it's important to note that N, N-dimethylaniline has a slightly lower pKa than pyridine. As a result, protonation takes place at the sp2 ring nitrogen.
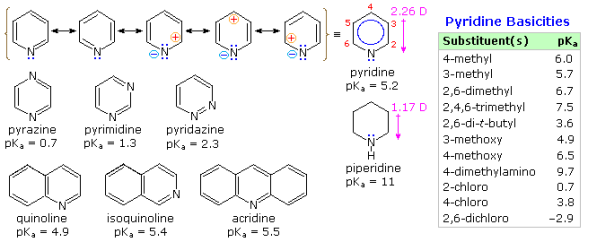
Because of the inductive effect of the second nitrogen, the diazines pyrazine, pyrimidine, and pyridazine are all weaker bases than pyridine. The order of base strength, on the other hand, is unexpected. The disparity between pyrazine and pyrimidine can be explained by considering the polar contributors, but the basicity of pyridazine appears to be unusual. The neutral base is thought to be destabilised in relation to its conjugate acid due to electron pair repulsion involving the vicinal nitrogens.
Electrophilic Substitution of Pyridine
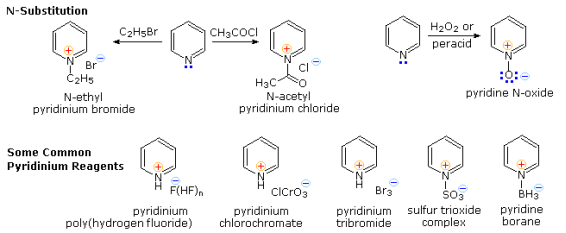
Pyridine is a relatively weak base (pKa=5.2). Pyidinium species formed by N-substitution retain the aromaticity of pyridine because the basic unshared electron pair is not part of the aromatic sextet, as it is in pyrrole. In the absence of water or other reactive nucleophiles, N-alkylation and N-acylation products can be prepared as stable crystalline solids, as shown below. N-acyl salts can be used as acyl transfer agents when making esters and amides. The pyridinium cation has been used as a moderating factor in complexes with a variety of reactive inorganic compounds due to its stability. At the bottom of the diagram, there are some examples of these safe and easy-to-handle reagents. The tribromide salt is a convenient source of bromine, and the poly (hydrogen fluoride) salt is a convenient source of HF for addition to alkenes and conversion of alcohols to alkyl fluorides. Pyridinium chlorochromate (PCC) and its associated dichromate analogue are versatile oxidation agents, and the tribromide salt is a convenient source of HF for addition to alkenes and conversion of alcohols to alkyl fluor Similarly, the reactive compounds sulphur trioxide and diborane can be treated as pyridine complexes with ease and safety.
As shown in the upper right equation, amine oxide derivatives of 3o-amines and pyridine are easily prepared by oxidation with peracids or peroxides. Treatment with zinc (or other reactive metals) in dilute acid normally results in reduction back to the amine.
We expect pyridine to undergo electrophilic substitution reactions much more slowly than benzene, based on the previous resonance explanation. Furthermore, the electrophilic reagents and catalysts used in these reactions coordinate with the nitrogen electron pair, exacerbating the positive charge at positions 2,4, and 6 of the pyridine rings, as shown above by clicking on the diagram. On the left are three examples of the extreme conditions needed for electrophilic substitution. As shown in the examples in the blue-shaded box at the lower right, substituents that block electrophile coordination with nitrogen or reduce the basicity of the nitrogen promote substitution, but substitution at C-3 remains dominant. The ease and regioselectivity of substitution are also influenced by activating substituents at other locations. Three examples will appear on the left after clicking on the diagram a second time. The amine substituent in the upper case guides the substitution to C-2, but the middle example's weaker electron-donating methyl substituent cannot resolve the propensity for 3-substitution. As seen in the bottom left for the 2-isomer, hydroxyl substituents at C-2 and C-4 tautomerize to pyridones.
Some electrophilic substitutions occur at C-4, while others occur at C-3 in pyridine N-oxide. As seen in the two examples on the right, the coordinate covalent N–O bond can exert a push-pull effect. Although the positively charged nitrogen alone has a strong deactivating effect, by -bonding to the ring nitrogen, the negatively charged oxygen will add electron density at C-2, C-4, and C-6. This is a limiting factor in the relatively facile nitration at C-4. If the oxygen is bound to an electrophile, such as SO3, the pyridinium ion that results reacts slowly and preferentially at C-3.
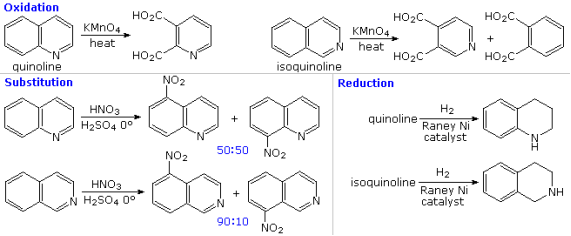
The fused ring heterocycles quinoline and isoquinoline contribute to the proof for the pyridine ring's stability. Quinoline undergoes vigorous permanganate oxidation, which primarily attacks the benzene ring; isoquinoline yields products from cleavage of both rings. It's worth noting that naphthalene is oxidised to phthalic acid in the same way. The heterocyclic ring in both compounds, on the other hand, is preferentially catalytically hydrogenated to produce tetrahydroproducts. In agreement with the preceding definition of similar pyridine reactions and the kinetically preferred substitution of naphthalene at C-1 () rather than C-2 (), electrophilic nitration, halogenation, and sulfonation typically take place at C-5 and C-8 of the benzene loop.
Other Reactions of Pyridine
Pyridine compounds are more susceptible to nucleophilic substitution reactions than similar benzene derivatives due to the nitrogen in the ring. Reaction 1 depicts the replacement of a 2-chloro substituent by an ethoxide anion in the following diagram. The ability of nitrogen to support a negative charge aids the addition-elimination process shown for this reaction. While a similar intermediate may be written for the substitution of a 4-halopyridine, the lack of such an intermediate prevents substitution at the 3-position. The leaving anion in reactions 2 and 3 of the Chichibabin aminations is hydride, which is unusual (or an equivalent). During these reactions, hydrogen is frequently produced. Quinoline is aminated at both C-2 and C-4 according to this process.
As shown by reactions 4 and 5, adding strong nucleophiles to N-oxide derivatives of pyridine proceeds more quickly than adding strong nucleophiles to pyridine itself. By removing the –OM substituent on nitrogen, the dihydro-pyridine intermediate quickly loses water or its equivalent.
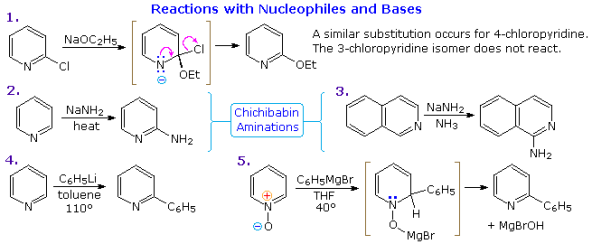
Five more examples of base or nucleophile reactions with substituted pyridine will be shown if you click on the diagram above. Alkyl substituents in the 2- and 4-locations are activated in the same way as carbonyl groups because the pyridine ring (and to a lesser extent the N-oxide ring) can support a negative charge. Alkylation and condensation reactions result from this activation, as seen in reactions 6 and 7. Reaction 8 shows how to make N-alkylpyridine by adding hydroxide to a N-alkyl pyridinium cation and then mildly oxidising it. Birch reduction transforms pyridines to bis-enamine dihydropyridines, which can then be hydrolyzed to form 1,5-dicarbonyl compounds. Pyridinium salts undergo a one-electron transition, resulting in exceptionally stable free radicals. The example in reaction 9 is a distillable green liquid that is stable (in the absence of oxygen). The strong base sodium amide causes amination through a pyridine intermediate, despite the fact that 3-halopyridines do not undergo addition-elimination substitution reactions like their 2- and 4-isomers. Reaction 10 is an example of this. It's important to note that 3-pyridyne is formed before 2-pyridyne. If C-4 is occupied by an alkyl substituent, the latter is formed. The pyridine intermediate has benzyne-like properties.
3.1.4 Some Polycyclic Heterocycles
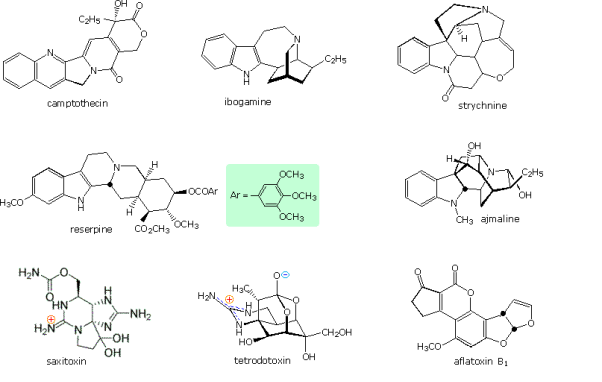
Many natural products contain heterocyclic structures. In the amine portion, examples of some nitrogen compounds known as alkaloids due to their basic properties were given. In the diagram below, you'll see some more examples. Camptothecin is a quinoline alkaloid that inhibits topoisomerase I, a DNA enzyme. Reserpine is an indole alkaloid that has traditionally been used to treat high blood pressure and psychotic activity. Strychnine and ajmaline are both indole alkaloids, with the former acting as an antiarrhythmic and the latter as a highly toxic pesticide. The neurotoxins saxitoxin and tetrodotoxin are both marine-derived neurotoxins of guanidiniun moieties. The Aspergillus fungus produces a non-nitrogenous carcinogenic compound called aspertoxin B1.
Porphyrin is a cyclic tertrapyrrole that is found in the heart of heme and chlorophyll. By clicking on the diagram, these structures will be drawn above.
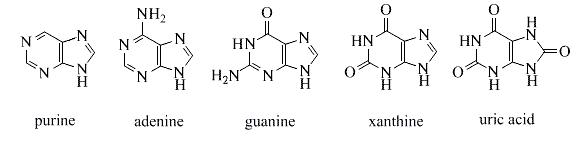
Natural products derived from the simple fused ring heterocycle purine are a particularly significant and abundant group. The amino acids adenine and guanine are two complementary bases that are needed for DNA to function. The structures of these compounds are depicted in the diagram below. The metabolic oxidation of purines produces xanthine and uric acid. Uric acid is usually excreted in the urine; however, an excess of uric acid in the bloodstream may cause gout, an arthritic disorder.
By clicking on the diagram above, examples of typical methylated purines will be drawn. The most well-known of these is caffeine, a bitter, crystalline alkaloid. It's present in different amounts in the beans, leaves, and fruit of some plants, along with other alkaloids including the cardiac stimulants theophylline and theobromine. Caffeinated beverages, such as coffee, tea, and some soft drinks, are arguably the most commonly consumed beverages on the planet. Caffeine is a central nervous system stimulant that helps people stay awake and alert. Caffeine's main metabolite in the body is paraxantheine.
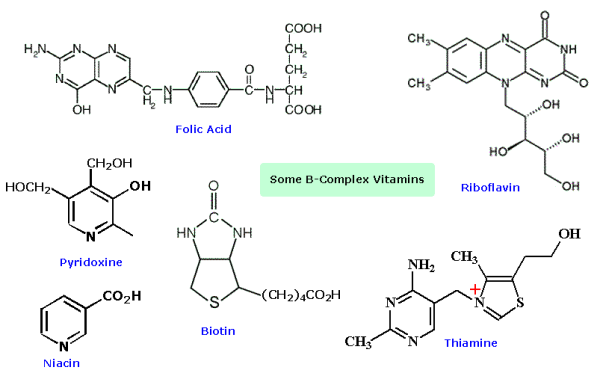
Sulfur heterocycles occur naturally, but to a lesser extent than their nitrogen and oxygen counterparts. Biotin and thiamine, two members of the B-vitamin family, contain heterocyclic moieties. In the diagram below, these are shown alongside other heterocyclic B-vitamins.
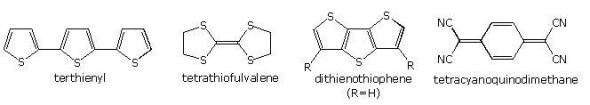
Terthienyl is a thiophene trimer that is present in the roots of marigolds and has nemicidal properties. Many species are phototoxic when exposed to UV irradiated terthienyl, according to studies. Polymers with thiophene groups, as well as fused structures like dithienothiophene, have intriguing electromagnetic properties and may be used as organic metal-like conductors and photovoltaic materials. Tetrathiofulvalene and tetracyanoquinodimethane form a charge transfer complex with one of the highest electrical conductivities ever recorded for an organic solid.
Key takeaway:
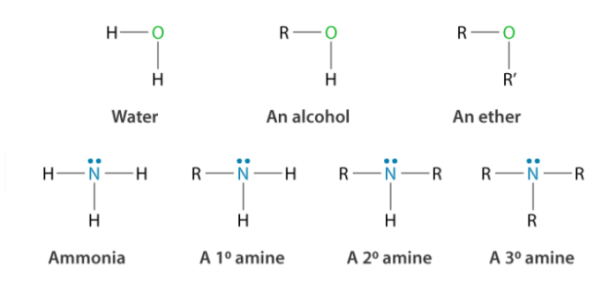
3.2.1 Amines:
Amines are a class of organic compounds that are formed by replacing one or more hydrogen atoms in a molecule of ammonia with an alkyl or aryl group. Vitamins, proteins, hormones, and other substances contain them. They're used in the production of a wide range of drugs and detergents.
3.2.2 Classification of amines:
Amines are graded as primary (1o), secondary (2o), or tertiary (3o) depending on how many hydrogen atoms are substituted by an alkyl or aryl group in ammonia (3o). Amines of the type R-NH2 or primary amines (1o) are obtained when only one hydrogen atom is substituted. Secondary amines are formed when two of the three hydrogen atoms are replaced by alkyl/aryl groups. Tertiary amines are formed when all three hydrogen atoms are replaced by an alkyl/aryl ring.
3.2.3 Nomenclature:
The names of compounds that are globally recognised in organic chemistry are given according to the IUPAC guidelines for organic compound nomenclature. Aliphatic amines are named by adding an alkyl group to the end of the amine, so their names are in the form of alkylamine. Methylamine is the name given to CH3NH2 (alkyl component + amine = methylamine). If two or more equivalent groups are present, prefixes such as di and tri are added before the names of the alkyl groups. If the amine contains more than one amino group, the parent chain and amino group positions are determined by counting the carbon atoms in the parent chain. The carbon atoms with the –NH2 groups are given the lowest numbers in the numbering system. The number of amino groups and their position in the molecule are then denoted by prefixes and numbers. Ethane 1, 2-diamine, for example, is designated as H2N-CH2-CH2-NH2.
Arylamines are formed when a –NH2 group is added to a benzene bond.
C6H5NH2 is one of the most basic examples of arylamine. It's also known as aniline, which is a recognised IUPAC name. When we call arylamines according to the IUPAC guidelines, the arene's e is substituted by amine, for example, C6H5-NH2 is called benzenamine.
3.2.4 Haloalkane - Classification and Nomenclature:
Haloalkanes, also known as alkyl halides, are a class of chemical compounds in which one or more hydrogen atoms have been substituted by a halogen atom (fluorine, chlorine, bromine, or iodine). The structural and physical properties of haloalkanes differ significantly from the structural and physical properties of alkanes. The structural variations are caused by the substitution of one or more hydrogen atoms with a halogen atom, as previously stated. Electronegativity, bond length, bond strength, and molecular size are all factors that influence physical properties. The following are a few examples of alklyl halides.
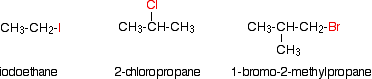
For multi-step organic synthesis, alkyl halides are a versatile and useful functional group. The structural classifications of alkyl halides can be used to predict their reactivity. The terms major, secondary, and tertiary are used to describe the three distinct systems. The number of carbons bound to the carbon containing the halide determines the classification. This classification strategy is similar to that used for alcohols, and it is described in more detail below.
3.2.5 Classification of Alkyl Halides:
The bonding patterns of the atoms involved are used to classify functional groups. Halogens have only one neutral bonding pattern (three lone pairs and a single bond), so their classification cannot be determined by them. The bonding pattern of the carbon bound to the halogen is used to assess the classification of alkyl halides, as shown in the diagram below.
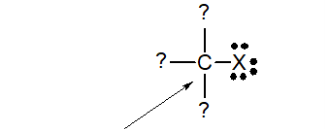
Alkyl halide classification is determined by the carbon bonded to the halogen
Primary alkyl halides
The carbon carrying the halogen atom is only bound to one other alkyl group in a primary (1°) halogenoalkane. The following are some examples of primary alkyl halides:

It's worth noting that the complexity of the attached alkyl group has no impact. There is only one connection to an alkyl group from the CH2 group that holds the halogen in each case. Even if there are no alkyl groups attached to the carbon with the halogen on it, CH3Br and the other methyl halides are often counted as primary alkyl halides.
Secondary alkyl halides
The carbon with the halogen attached is joined directly to two other alkyl groups, which may be the same or different, in a secondary (2°) halogenoalkane. Consider the following examples:
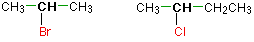
Tertiary alkyl halides
The carbon atom carrying the halogen is directly connected to three alkyl groups in a tertiary (3°) halogenoalkane, which can be any mixture of same or different. Consider the following examples:

3.2.6 IUPAC and Common Nomenclature
Since an alkyl halide (or haloalkane) is formed by replacing only one hydrogen atom, the nomenclature scheme is very similar to that for alkanes. The common names of alkyl halides are made up of two parts: the alkyl group's name and the stem of the halogen's name, with the suffix -ide.
Format of Common Name: alkyl name + halide name
The IUPAC scheme uses the parent alkane's name with a prefix indicating the halogen substituents, followed by a number indicating the position of the substituent. Fluoro-, chloro-, bromo-, and iodo- are the prefixes. Ethyl chloride is the common name for CH3CH2Cl, while chloroethane is the IUPAC name. The three sections of the IUPAC name for simple halo alkanes are shown below.
IUPAC Name Format: locator # + parent alkane + halo prefix
Popular names are given to alkyl halides with simple alkyl groups (one to four carbon atoms). IUPAC names are normally assigned to those with a greater number of carbon atoms.
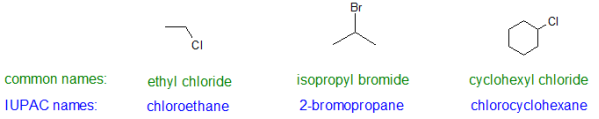
EXAMPLE:
Give each compound's common and IUPAC names.
1. CH3CH2CH2BrBrBrBrBrBrBrBrBr
2. (CH3)2CHCl (CH3)2CHCl (CH3)2CHCl (
the response
3.3. Structure, aromaticity in 5-membered and 6-membered rings containing one heteroatom:
3.3.1 Five-membered ring containing one heteroatom:
The structures of the parent aromatic compounds in this family—pyrrole, furan, and thiophene—are seen.
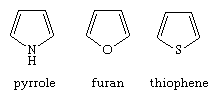
Pyrolidine, tetrahydrofuran, and thiophane are the saturated derivatives, respectively. The bicyclic compounds indole (or isoindole), benzofuran, and benzothiophene are made up of a pyrrole, furan, or thiophene ring fused to a benzene ring, respectively.
The nitrogen heterocycle pyrrole, as stated in the introduction, is found in bone oil and is produced by the decomposition of proteins when heated to high temperatures. The amino acids proline and hydroxyproline, which are components of several proteins and are contained in especially high amounts in collagen, the structural protein of bones, tendons, ligaments, and skin, contain pyrrole rings.
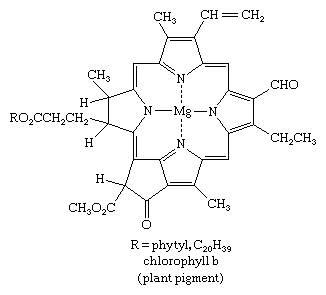
Pyrrole derivatives can be found all over the living world. Alkaloids, a broad class of alkaline organic nitrogen compounds formed primarily by plants, include pyrrole compounds. The most well-known pyrrole-containing alkaloid is nicotine. The heme group of the oxygen-carrying protein haemoglobin and associated compounds such as myoglobin; the chlorophylls, which are light-gathering pigments found in green plants and other photosynthetic organisms; and vitamin B12 are all made up of four pyrrole units linked together in a larger ring structure known as a porphyrin, such as chlorophyll b, shown below.
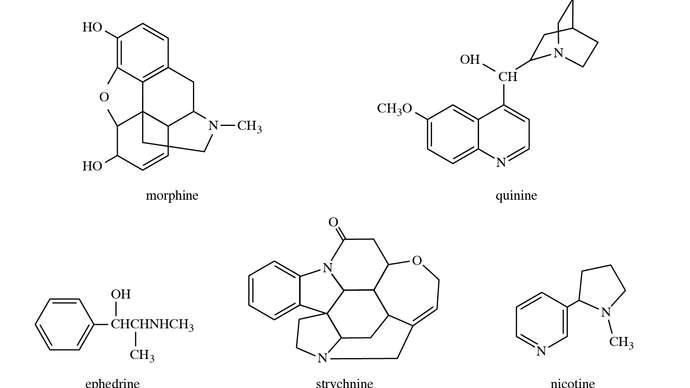
3.3.2 alkaloid
The alkaloids morphine, quinine, nicotine, strychnine, and ephedrine have chemical structures that are well-known.
Bile pigments are made up of a chain of four pyrrole rings that are formed as the porphyrin ring decomposes. The end product of the breakdown of heme from damaged red blood cells is bilirubin, the brownish yellow pigment that gives faeces its distinctive colour.
The phthalocyanines are a class of synthetic pigments made up of four isoindole units connected by a wide ring. Phosphocyanine blue is a common member of the family (Monastral Fast Blue).
One or more indole units can be found in the molecular structure of many compounds formed by plants and animals. The essential vat dye indigo, which contains two indole units, has been used for thousands of years and was previously derived from plants, but it is now synthetically produced on a large scale.
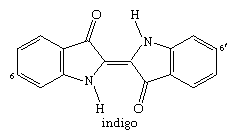
The two are closely related. Tyrian purple is 6,6′-dibromoindigo, a dye derived from snail organisms and used in classical times (with bromine atoms bonded to the numbered carbons in the structure above).
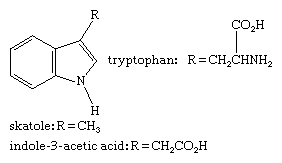
The body uses tryptophan, an indole-containing basic amino acid contained in most proteins, to make many important substances, including serotonin, a neurotransmitter, and niacin, a B-complex vitamin (see below). Rings of six members and one heteroatom). Skatole, a tryptophan degradation product that preserves the indole unit, is a major contributor to the heavy odour of mammalian faeces. The most important member of the auxin family of plant hormones, indole-3-acetic acid (heteroauxin or -indolylacetic acid), is a plant growth regulator. These compounds have the following structures:
The indole alkaloids, which have been isolated from plants belonging to more than 30 families, are perhaps the most well-known indole-containing compounds. This group includes the mushrooms psilocin and psilocybin, the ergot fungus alkaloids, the medicines reserpine and yohimbine, and the poison strychnine.
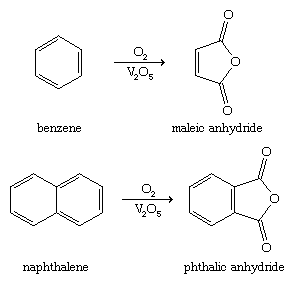
Furan, the most basic member of the furan family of oxygen heterocycles, is hydrogenated to produce tetrahydrofuran in the industrial world. Tetrahydrofuran is a solvent that is often used to make adipic acid and hexamethylenediamine, which are the raw materials for nylon-6,6, the most common form of nylon. Maleic anhydride and phthalic anhydride, which are components of resins and plastics, are two other furan derivatives of industrial interest. As shown, these compounds are made in bulk by oxidising benzene and naphthalene, respectively (V2O5 is a vanadium catalyst).
One or more simple sugar (monosaccharide) units make up all carbohydrates, the biochemical family that includes sugars and starches. These sugars are polyhydroxy aldehydes or polyhydroxy ketones, which occur as equilibrium mixtures of open-chain and cyclized forms in aqueous solution. The cyclized form of the sugar is often a furanose, a five-membered tetrahydrofuran ring, as shown below for fructose, or fruit sugar, as a cyclized isomer (called a fructofuranose).
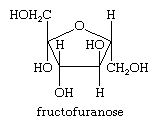
The sugars ribose and deoxyribose, which are present in furanose form in RNA and DNA, respectively, the heredity-controlling components of all living organisms, are other essential examples of tetrahydrofuran ring systems.
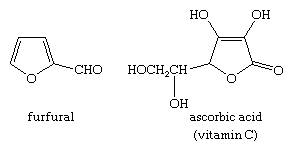
Furan derivatives are formed when certain carbohydrates are dehydrated. The conversion of a carbohydrate found in corncobs, oat husks, and other agricultural waste into furan-2-aldehyde, or furfural, which is widely used as a solvent, in the manufacture of plastics, and in the preparation of other furan derivatives, is of great commercial importance. Many other furan derivatives, including vitamin C, are found in nature. Furfural and vitamin C have the following structures:
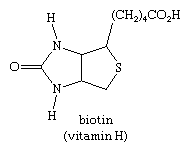
Coal tar and crude petroleum contain the sulphur heterocycle thiophene and associated compounds. Biotin, a B-complex vitamin, is the most essential biologically occurring thiophene derivative.
3.3.3 Six-membered rings with one heteroatom
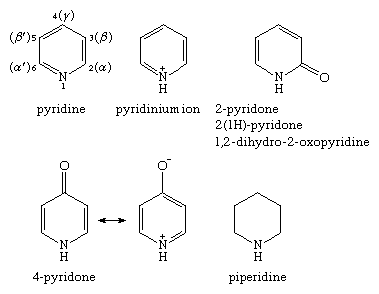
The following is the nomenclature for the various monocyclic nitrogen-containing six-membered ring compounds. The positions of pyridine on the ring are shown, with Arabic numerals preferred over Greek letters, though both systems are used. Since contributions to the resonance hybrid from charged resonance forms, such as that seen for 4-pyridone, the pyridones are aromatic compounds.
Picolines, lutidines, and collidines are mono-, di-, and trimethylpyridines—that is, pyridines with one, two, or three attached methyl groups, respectively—with the position of the methyl groups denoted by numbers—e.g., 2,4,6-collidine. Picolinic, nicotinic (derived from nicotine, of which it is an oxidation product), and isonicotinic acid are all common names for pyridine-2-, -3-, and -4-carboxylic acids. Coal tar and bone oil contain pyridine, as well as picolines, lutidines, and collidines. Pyridine derivatives are also very important in biology. For example, nicotinic acid is more generally known as the B-complex vitamin niacin; nicotinamide, or niacinamide, is a nutritionally similar type of niacin. Pyridoxine, or vitamin B6, is another member of the B complex. Pyridoxine and nicotinamide have the following structures:
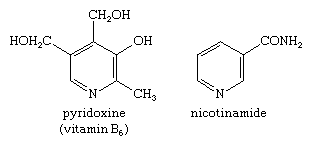
Nicotinamide adenine dinucleotide (NAD, also called coenzyme I) and nicotine adenine dinucleotide phosphate (NADP, coenzyme II) are two coenzymes derived from nicotinamide, and pyridoxal phosphate (codecarboxylase) is a physiologically active type of pyridoxine. Many alkaloids, including nicotine (mentioned in the previous section for its pyrrole ring) and piperine (one of the sharp-tasting constituents of white and black pepper, from the plant species Piper nigrum), have pyridine or piperidine ring structures, as shown.
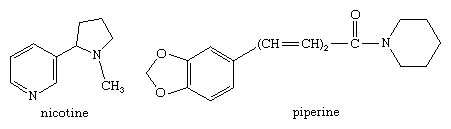
Pyridine is an essential solvent and intermediate used to produce other compounds. It was once extracted commercially from coal tar but is now made catalytically from tetrahydrofurfuryl alcohol and ammonia. Vinylpyridines, for example,
Completely saturated pyridine, piperidine, is used in rubber manufacturing and as a chemical raw material, and is an effective monomer building block for plastics.
The tuberculostat isoniazid (isonicotinic acid hydrazide), the anti-AIDS medication nevirapine, the vasodilator nicorandil, the urinary-tract analgesic phenazopyridine, and the anti-inflammatory sulfa drug are all examples of pharmaceutically essential pyridines. Piperidine (1-phenylcyclohexyl) (PCP, phencyclidine) was developed as an anaesthetic, but its potent hallucinogenic effects have led to violence. Herbicides such as diquat, paraquat, clopyralid, and diflufenican are pyridine derivatives.
Two isomeric benzopyridines (upper pair) and two isomeric dibenzopyridines (lower pair) are shown in the structural formulas below, along with their common names and agreed numberings. Coal tar was used to make all four compounds, as well as some of their alkyl derivatives. Each one is also the parent substance of an alkaloid class. Quinolines (e.g., quinine and other derivatives still derived from the Cinchona tree) and isoquinolines (e.g., morphine) are the most well-known of these groups.
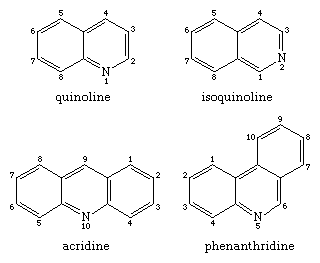
Quinoline can be used to make nicotinic acid as well as other compounds like drugs and dyes. Synthetic quinoline activity far outnumbers that of coal tar. Morphine, codeine, and thebaine are alkaloids of the opium poppy that contain partially reduced isoquinoline rings and have been used as hypnotics and analgesics for decades. Heroin, a semisynthetic derivative of morphine, is a much more hypnotic and highly addictive substance.
Cyanine dyes, which are used as sensitizers in silver halide photographic emulsions, and quinophthalone dyes, which are used in plastics, polymer textiles, paper, cosmetics, transfer-printing processes, electronic imaging, and laser dyes, are important synthetic derivatives of the benzo- and dibenzopyridines. Antiseptics such as the coal-tar dye acriflavine, antimalarial agents such as mepacrine (quinacrine) and chloroquine, antibacterial agents such as ciprofloxacin, and trypanocides (drugs that kill trypanosomes, parasitic protozoans responsible for Chagas disease, sleeping sickness, and other serious infectious diseases) are among the other derivatives.
The parent six-membered, aromatic, monocyclic oxygen and sulphur compounds of pyrylium and thiopyrylium are positively charged ions (cations) of their respective groups.
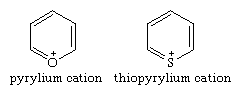
Only if the ring contains a carbonyl group (i.e., a ring carbon atom connected by a double bond to an oxygen atom not in the ring) is an uncharged aromatic (completely conjugated) six-membered ring containing an oxygen or sulphur atom possible, as in the pyrones.
The pyrans have extra hydrogen atoms, which are shown by a number followed by a H in structural diagrams. Certain sugars, such as the monosaccharide glucose, are referred to as pyranoses because they contain six-membered tetrahydropyran rings with the following structure:
Pyrone derivatives can be found in a variety of natural products. For example, kojic acid is an antibiotic produced by the action of moulds on starches or sugars. Bufotoxin, a toxic ester of the steroid bufotalin, is derived from the skin glands of toads (genus Bufo; see steroid: Structural relationships of the principal categories of steroids).
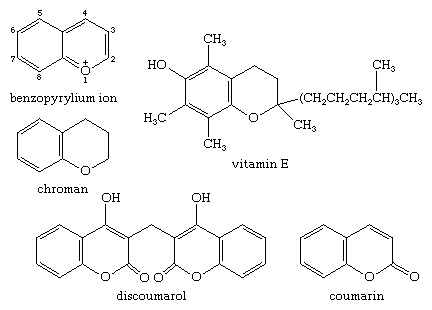
A significant number of natural products are derived from the benzopyrylium cation. The chroman unit, also known as 3,4-dihydro-2H-1-benzopyran, is not found in nature, but it is present in many natural products. Vitamin E (-tocopherol), a substituted chroman, is present in plant oils and green vegetable leaves, while coumarin, or 2H-1-benzopyran-2-one, used in perfumes and flavourings, and its derivative dicoumarin (dicumarol, or discoumarol), a blood anticoagulant, are both living organisms' products.
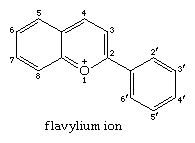
The flavylium cation is the parent of anthocyanidines, which combine with sugars to form anthocyanin pigments, which are the natural red and blue colourants found in flowers and fruits. Anthoxanthins, a subgroup of flavonoids, have a colour spectrum ranging from yellow to reddish orange. The above are flavone (2-phenyl-4-pyrone) derivatives with hydroxy or methoxy groups replacing one or more hydrogen atoms. The composition of the flavylium ion is as follows:
3.4.1 Paal-Knorr Furan Synthesis
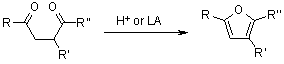
One of the most common methods for the preparation of furans is the acid-catalyzed cyclization of 1,4-dicarbonyl compounds, known as the Paal-Knorr synthesis. The Paal-Knorr reaction's synthetic utility has increased as a result of the recent development of numerous methods for the synthesis of 1,4-diones.
Mechanism of the Paal-Knorr Furan Synthesis
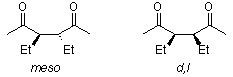
The cyclization rates of meso- and dl-3,4-diethyl-2,5-hexanediones were compared, and it was discovered that the stereochemical structure of the unchanged dione was retained during the reaction. These results contradict the widely accepted mechanism, which requires the ring closure of a rapidly formed monoenol, as seen here.
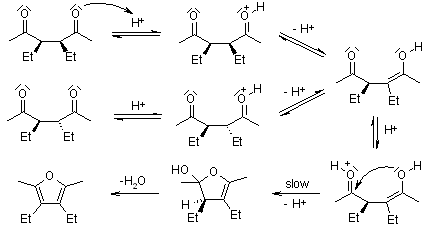
The rate of acid-catalyzed enolization is considered to be unaffected by the ketone's structure. The variations in reaction rate cannot be explained by this mechanism since the rate-determining phase is the same for both substrates.
Below is a diagram of how the substituents will interact differently in the rate-determining phase. The ease with which both molecules can achieve a suitable conformation for cyclization is not the same:
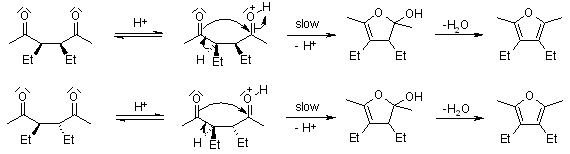
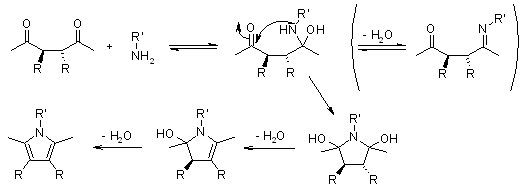
3.4.2 Paal-Knorr Pyrrole Synthesis
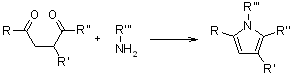
The Paal-Knorr Pyrrole Synthesis produces pyrrole by combining a 1,4-dicarbonyl compound with an excess of a primary amine or ammonia.
Under neutral or weakly acidic conditions, the reaction can be carried out. The use of a weak acid, such as acetic acid, speeds up the reaction, but amine/ammonium hydrochloride salts or reactions at pH 3 result in furans as the main product (Paal-Knorr Furan Synthesis).
Mechanism of the Paal-Knorr Pyrrole Synthesis
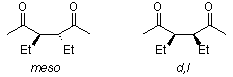
Venkataraman Amarnath demonstrated that meso- and dl-3,4-diethyl-2,5-hexanediones cyclize at different rates and that the stereochemical structure of the unchanged dione is retained during the reaction (J. Org. Chem., 1991, 56, 6924). Any process involving the creation of an enamine prior to the rate-determining phase of cyclization, such as the one below, must be ruled out.
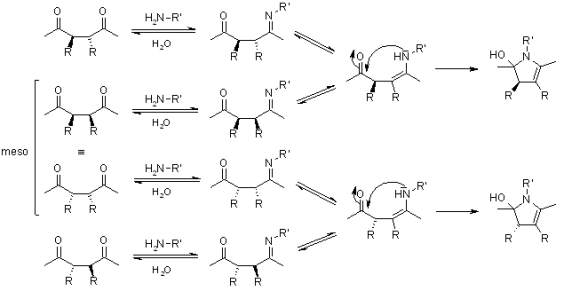
A charged immonium ion must be an intermediate if the ring is produced from an imine created from a primary amine. Amarnath used various aryl groups as substituents to stabilise or destabilise the immonium ion:
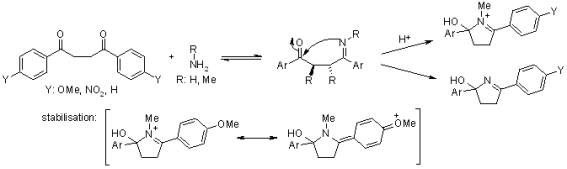
The use of ammonia can result in an uncharged intermediate, which is less influenced by the substitutents used. The basicity of the imine is also influenced by the substituents, with the nitro group resulting in a more specific nucleophile. Ammonia and methylamine were used to compare cyclization rates. The nitro group has had a positive impact on the reaction rate in any case. In each case, the methoxy group has a negative effect on the cyclization rate. For a potential mechanism involving an immonium ion, a comparison of the relative reaction rates of all substrates (R: H, Me) revealed no clear stabilization/destabilization effect.
The cyclization of a hemiacetal, accompanied by different dehydration steps, is a process that accounts for the effect of different substitution patterns (meso, dl) and explains the influence of a p-nitrophenyl group making a nucleophile more reactive (but not as the imine):
Key takeaway:
3.5.1 Hantzsch Dihydropyridine (Pyridine) Synthesis
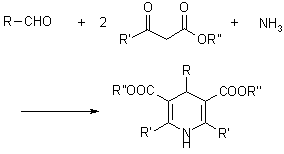
The condensation of an aldehyde with two equivalents of a -ketoester in the presence of ammonia makes the preparation of dihydropyridine derivatives. Following oxidation (or dehydrogenation), pyridine-3,5-dicarboxylates are formed, which can then be decarboxylated to yield pyridines.
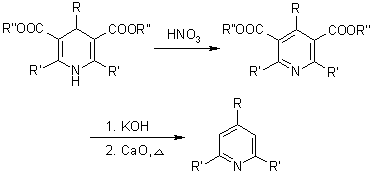
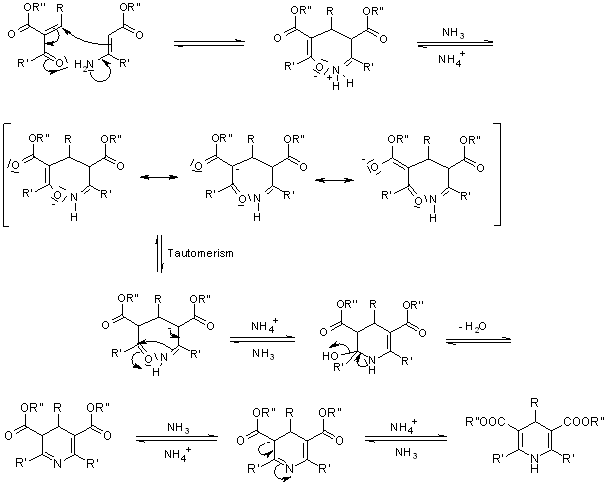
Mechanism of the Hantzsch Dihydropyridine Synthesis
As a main intermediate, the reaction can be visualised as passing through a Knoevenagel Condensation product:
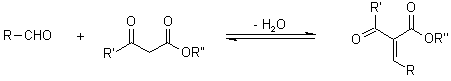
The ester enamine, which is formed by condensation of the second equivalent of the -ketoester with ammonia, is a second important intermediate:
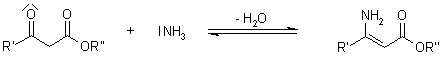
The dihydropyridine derivative is formed by further condensation between these two fragments:
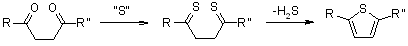
3.5.2 Paal Thiophene Synthesis
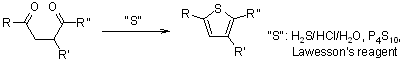
The Paal-Knorr Thiophene Synthesis involves the condensation of a 1,4-dicarbonyl compound in the presence of an excess of a sulphur source, such as phosphorous pentasulfide or Lawesson's reagent, to produce thiophenes.
Attention: regardless of the sulphur source, some toxic H2S forms as a by-product.
Mechanism of the Paal-Knorr Thiophene Synthesis
Phosphorus pentasulfide or Lawesson's reagent function as both sulfurizing and dehydrating agents, allowing for a reaction pathway that could lead to the formation of furans first. Foye (J. Org. Chem., 1952, 17, 1405) tested this hypothesis by treating various 1,4-dicarbonyl compounds and their possible furan intermediates (such as acetonylacetone and 2,5-dimethylfuran) with phosphorus pentasulfide. The disparities in 2,5-dimethylthiophene yields under the same reaction conditions rule out the possibility of a dominant reaction pathway involving furan intermediates:
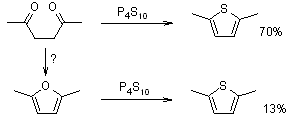
Foye suggested the following reaction pathway:

The presence of a bis-thioketone intermediate is now thought to be possible but not needed (J. Schatz, Science of Synthesis, George Thieme Verlag Stuttgart, 2000, Vol. 9, 298.)
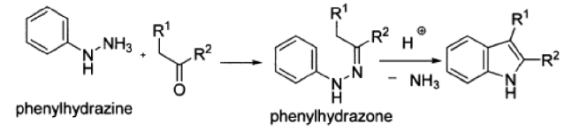
3.6.1 What is Fischer Indole Synthesis?
Both organic and medical chemists and medicinal chemists have found enough opportunities in indole synthesis. Thousands of indole derivatives are generated each year in the quest for life-saving drugs.
Fischer invented the Fischer indole synthesis in 1883, and it is one of the oldest and most powerful methods of indole growth. In the presence of Brnsted or Lewis acids, a number of indoles may be made from aryl hydrazines and substituted ketones or aldehydes.
The Fischer indole synthesis, in which an aromatic phenylhydrazone is heated in acid, is the most useful route to indoles. The condensation product of a phenylhydrazine and an aldehyde or ketone is phenylhydrazone. A cyclic rearrangement mechanism is used to close the ring.
3.6.2 Fischer Indole Synthesis Reaction:
Since Emil Fischer's discovery of the Fischer indole synthesis in 1883, it has become the most widely used method for preparing indole rings. In essence, the Fischer indole synthesis is the cyclization of an arylhydrazone, which is formed by treating an aryl hydrazine with an aldehyde or a ketone with an acid catalyst or by heating it to form the indole nucleus. For over a century, scientists have been studying the process in depth, and several intermediates have been isolated and characterised.
Below is the Fischer indole synthesis reaction.
Indole synthesis has possibly received more attention than any other single heterocyclic method, and as a result, several routes are available; ring syntheses of benzo furans and benzo thiophenes, on the other hand, have received much less attention. The Fischer indole synthesis, which has been around for over a century, is still commonly used: an aryl hydrazone is heated with an acid, a multi-step sequence follows, ammonia is lost, and an indole is generated.
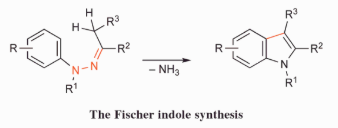
The synthesis is often carried out by directly indolizing an equimolar mixture of aryl hydrazine and aldehyde or ketone without first isolating the hydrazone. Similarly, aryl hydrazones prepared by reduction of the corresponding aryldiazonium salt or N-nitroso arylalkylamine or by a palladium-mediated coupling reaction can be indolized directly in the presence of the carbonyl moiety without isolation of the aryl hydrazone. When the aryl hydrazone intermediates are unstable or toxic, such methods are useful.
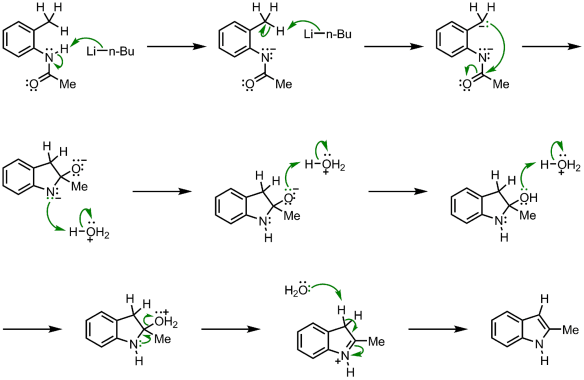
3.6.3 Fischer Indole Synthesis Reaction Mechanism
The enamine lautomer is formed by protonation and isomerization of the arylhydrazone formed by condensation of an aryl hydrazine and a carbonyl compound. The protonated enamine lautomer then undergoes an irreversible [3,3] – sigmatropic electrocyclic rearrangement, in which the N-N bond is broken.
The benzene ring is then re-aromatized by the resulting double imine, yielding an anilino imine, whose nucleophilic amine group attacks the imine intramolecularly to yield the amino indoline. The indole is generated after the loss of an ammonia molecule and aromatization.
As hydrazine reacts with a carbonyl compound, it behaves like an amine and produces a hydrazone, which is an imine-like substance. The enamine tautomer of this hydrazone undergoes the cycle rearrangement, which occurs because the cyclic movement of electrons creates a tight C-C bond thus cleaving a weak N-N bond. This seems to be the formation of a di-imine.
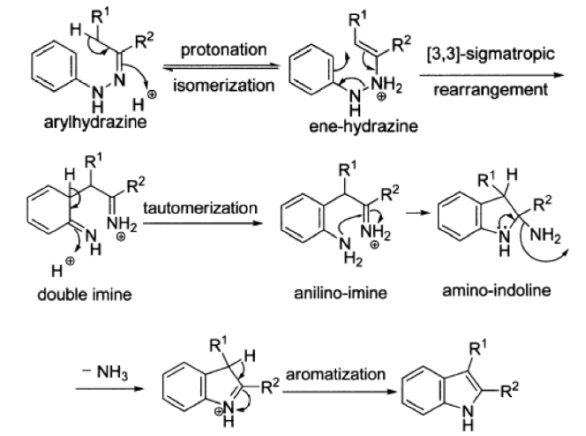
The rearomatization of one of these results in the formation of nan aromatic amine. The other imine feature is then attacked, yielding the nitrogen equivalent of a hemiketal. Finally, the aromatic indole system is formed by ammonia elimination catalysed by an acid.
3.6.4 Drawbacks of Fischer Indole Synthesis:
Unfortunately, the reaction fails when acetaldehyde is used, so it can't be used to make indole. Instead, you can use the keto acid pyruvic acid and decarboxylate the result to get indole.
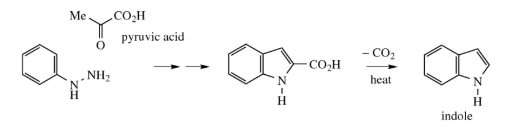
3.6.5 Features of Fischer Indole Synthesis
3.7.1 Furfural Acid:
Preparation:
Furfural may be obtained by the acid catalyzed dehydration of 5-carbon sugars (pentoses), particularly xylose.
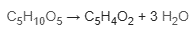
These sugars can be made from pentosans, which are found in lignocellulosic biomass as hemicellulose.
Depending on the form of feedstock, between 3% and 10% of the mass of crop residue feedstocks can be recovered as furfural. Furfural and water detach from the reaction mixture as they evaporate and condense. As of 2012, the global production capacity was about 800,000 tonnes. China is the world's largest supplier of furfural, with the majority of global capacity. Illovo Sugar in South Africa and Central Romana in the Dominican Republic are the other two major commercial producers.
Furfural can be made from plant material in the lab by heating it with sulfuric acid or other acids. A worldwide effort to replace sulfuric acid with easily-separable and reusable solid acid catalysts has been studied in order to prevent poisonous effluents.
After the furfural is removed in industrial processing, some lignocellulosic residue remains. This residue is dried and burned to generate steam for the furfural plant's activity. Excess residue from newer, more energy efficient plants is or can be used for co-generation of electricity, cattle feed, activated carbon, mulch/fertilizer, and other purposes.
3.7.2 Furoic Acid:
Preparation:
The oxidation of either furfuryl alcohol or furfural produces 2-furoic acid. This can be accomplished chemically or by biocatalysis. The Cannizaro reaction of furfural in an aqueous NaOH solution is currently the industrial path, which produces both 2-furoic acid and furfuryl alcohol. [5] The microorganism Nocardia corallina is involved in the bio-catalytic pathway. Experiments with this microbial conversion yielded high yields: 98 percent from 2-furfuryl alcohol and 88 percent from 2-furanaldehyde, respectively. Because most other microorganisms create two products from oxidation, the acid and the alcohol, N. corallina's oxidation is special. Furthermore, there is no aromatic ring destruction.
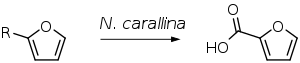
Biotransformation of furfuryl alcohol (R = CH2OH) or furfural (R = CHO) to 2-furoic acid by Nocardia corallina
Key takeaway:
References:
1. Graham Solomons T. W., Fryhle, Craig B., Snyder Scott A, Organic Chemistry, Wiley
Student Ed, 11th Edition (2013)
2. Jonathan Clayden, Nick Greeves, Stuart Warren, Organic Chemistry, 2nd Edition, Oxford
Publisher, 2014.
3. Dhawan, S.N., Pradeep’s Organic Chemistry, (Vol. I and II), Pradeep Publications




