Unit – 1
Organometallic Compounds – I
1.1.1 Definition:
Organometallic Compounds are chemical compounds which contain at least one bond between a metallic element and a carbon atom belonging to an organic molecule. Even metalloid elements such as silicon, tin, and boron are known to form organometallic compounds which are used in some industrial chemical reactions.
The catalysis of reactions wherein the target molecules are polymers or pharmaceuticals can be done with the help of organometallic compounds, resulting in an increase in the rate of the reactions.
Generally, the bond between the metal atom and the carbon belonging to the organic compound is covalent in nature. When metals with relatively high electropositivity (such as sodium and lithium) form these compounds, a carbanionic nature is exhibited by the carbon which is bound to the central metal atom.
An organometallic compound is one in which a carbon atom is bonded to a metal atom.
Above is an example of an organometallic compound in which the carbons of a benzene molecule bind with chromium. Grignard reagents, tetracarbonyl nickel, and dimethyl magnesium are a few additional examples of these types of chemicals.
1.1.2 Classification of Organometallic Compounds
Organometallic compounds are classified in three classes.
(i) Sigma bonded organometallic compounds: For example, in Grignard reagents, R – Mg – X, where R is an alkyl or aryl group and X is a halogen, the metal atom and the carbon atom of the ligand are bonded together with a sigma bond.
(b) Zinc compounds of the formula R2Zn, such as (C2H5)2Zn, (C2H (isolated by Frankland).
Other compounds that are comparable include (CH3)4Sn, (C2H5)4Pb, Al2(CH3)6, Al2(C2H5)6, Pb(CH3)4, and so on.
Al2(CH3)6, Al2(C2H5)6, Pb(CH3)4, (CH3)4Sn, (C2H5)4Pb, (CH3)4Sn, (C2H5)4Pb, (CH3)4Sn,
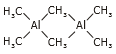
Al2(CH3)6 is a dimeric molecule with a structure that resembles diborane (B2H6). Two methyl groups operate as bridges between two aluminium atoms in this electron-deficient molecule.
(ii) Pi-bonded organometallic compounds: Metal compounds with alkenes, alkynes, benzene, and other ring compounds are known as alkenes, alkynes, benzene, and other ring compounds. The metal and ligand establish a connection in these complexes that involves the ligand's -electrons. Zeise's salt, ferrocene, and dibenzene chromium are three common examples. These are displayed below.
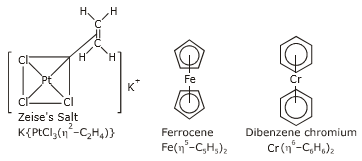
The number of carbon atoms bonded to the metal in these compounds is indicated by the Greek letter η(eta) with a number. The prefixes η2, η5 and η6 indicate that 2, 5 and 6 carbon atoms are the metal in the compound.
(iii) Sigma and Pi bonded organometallic compounds: This class includes metal carbonyl compounds, which are generated when metal reacts with carbon monoxide. Both - and -bonding are present in these compounds. In these compounds, the oxidation state of metal atoms is usually zero. Mononuclear, bridging, and polynuclear carbonyls exist.
Ni(CO)4 Fe(CO)5 Cr(CO)6 Fe(CO)9Ni(CO)4 Fe(CO)5 Cr(CO)6 Fe(CO)9
The metal-carbon bond in a metal carbonyl has both the and character. A as well as a personality. When a vacant hybrid orbital of the metal atom overlaps with an orbital on the C atom of carbon monoxide carrying a lone pair of electrons, a bond is created.
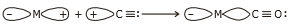
When a full orbital of a metal atom overlaps with a vacant antibonding* orbital of the C atom of carbon monoxide, a p-bond is formed. Back donation of electrons by metal atoms to carbon is another name for this overlap.
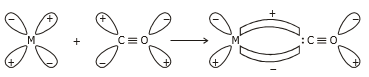
The -overlap is parallel to the -bond's nodal plane.
The bonding -orbital electrons in olefinic complexes are donated to the metal atom's vacant orbital while also being donated to the olefinic's back bonding p-orbital.
1.2.1 What is hapticity of an organometallic ligands:
The hapticity of an organometallic ligand is a straightforward concept: it simply indicates how many atoms in the ligand are linked to the metal centre. The Greek letter “eta” is commonly used in chemistry literature to represent this. A ligand with two atoms bound to the metal centre is called a 2, a ligand with one atom is called a 1, and so on. The most frequent hapticity for a ligand is 1, though ligands with higher coordination numbers, such as the cyclopentadienyl anion ligand (typically 5) are not uncommon.
Or
When a lone pair is bonded to a transition metal, the lone pair's atom forms a bond with the metal. Which atom is connected to the metal when a pi bond is formed? They're both of them. This bonding condition involves three atoms rather than just two. Both carbon atoms that make up the original pi bond are now donating it to the metal.
It may seem much more difficult to describe the way of binding when ligands are bound to a metal via a conjugated pi-system. When two double bonds appear in a succession, all four atoms that make up those two pi bonds donate to the metal. We can also think of this as one long pi bond because the bond is conjugated. All four donor atoms, as well as the metal atom, are involved in the pi bond to metal connection.
Consider the case of two independent pi bonds that aren't conjugated. A bidentate donor is a ligand that has two distinct pi bonds. Bidentate ligands bind to two donor locations in the cell. A ligand like 1,2-ethanediamine binds to both nitrogen atoms through their lone pairs. 1,5-hexadiene is thought to bind through the pi bond at both ends of the chain. Because all four carbons participate in connecting to the metal through one conjugated bond, 1,3-butadiene is a little different.
The phrase used to denote the simultaneous participation of several atoms. During The hapticity of pi coordination. A dihaptic donor is a normal alkene, such as ethene or propene, in which two carbons participate in the donation of one bond to the metal. A tetrahaptic donor is a conjugated alkene, such as 1,3-butadiene. A conjugated pi bond is donated to the metal by four carbon atoms. Of course, this conjugated diene has the ability to contribute four electrons at once, producing a double bond with the metal.
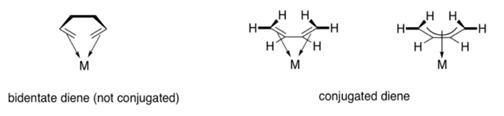
Figure: Multihaptic ligands that are commonly used.
The symbols 2 or 3 (read "eta-two" or "eta-three") in the figures above allude to the ligand's hapticity. A 2 ligand is dihaptic, with two atoms sharing the pi system's donation; a 3 ligand is trihaptic, with three atoms sharing the conjugated pi system's donation.
1.3.1 18 Electron rule:
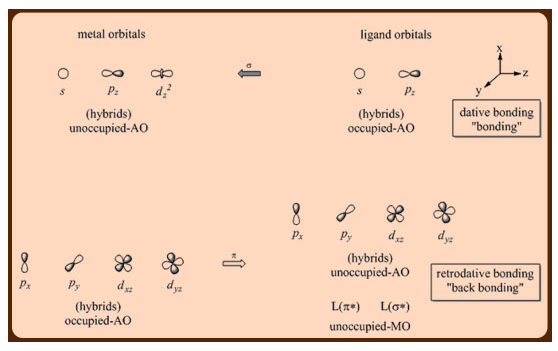
Transition metal organometallic compounds have a wide range of structural differences, which present themselves in a variety of chemical characteristics. The chemical catalysis potentials of several of these transition metal organometallic compounds are of primary interest. Unlike main group organometallic compounds, which primarily use the ns and np orbitals for chemical bonding, transition metal compounds frequently use the (n1)d, ns, and np orbitals (Figure 1). Due to the partial filling of these orbitals, these metal centres can operate as both electron donors and acceptors, allowing them to participate in -donor/-acceptor synergic interactions with donor-acceptor ligands such as carbonyls, carbenes, arenes, isonitriles, and so on.
The Inert Gas Rule, also known as the 18 Valence Electron (18 VE) Rule, or the Effective Atomic Number (EAN) Rule: The 18-valence electron (VE) rule asserts that thermodynamically stable transition metal complexes have 18 valence electrons, which include the metal d electrons as well as electrons supplied by metal bound ligands. The 18 valence electrons in transition metal complexes can be counted using either of the two electron counting methods: I the ionic approach, or (ii) the neutral approach. Please keep in mind that a metal-metal bond adds one electron to the metal atom's total electron count. One electron is donated by a bridging ligand to the bridging metal atom.
Ferrocene Fe(C5H5)2 is an example of a ferrocene.

Example 2. Mn2(CO)10
The majority of transition metal organometallic compounds fall into one of three categories.
1. Class I complexes with a valence electron count that deviates from the 18 VE rule.
2. Class II compounds with a valence electron count of no more than 18.
3. Class III compounds with valence electrons that follow the 18 VE rule to a tee.
The antibonding orbitals should not be occupied, but the nonbonding orbitals may be occupied while the bonding orbitals should be occupied, according to the guiding principle that determines the classification of transition metal organometallic complexes.
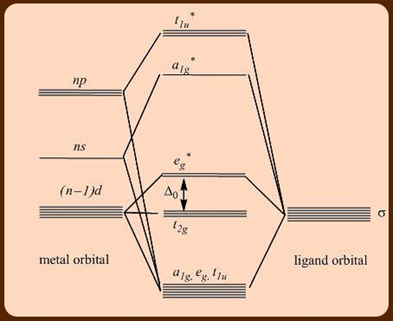
Figure: Interactions only molecular orbital diagram for an octahedral transition metal complex.

Figure: A simplified molecular orbital diagram for an octahedral transition metal complex, highlighting only the and interactions.
Class I:
The o splitting in class I complexes is minimal, and it frequently applies to 3d metals and ligands at the lower end of the spectrochemical series. The t2g orbital is nonbonding in this situation and could be inhabited by up to 6 electrons (Figure). 04 electrons can occupy the eg* orbital, which is weakly antibonding. As a result, this family of compounds might have a valence electron count of 1222. Tetrahedral transition metal complexes also fall into this category due to their low tetr splitting energy.
Class II:
The o splitting is relatively large in class II complexes, and it applies to 4d and 5d transition metals with high oxidation states, as well as ligands in the intermediate and higher ranges of the spectrochemical series. The t2g orbital in this situation is largely nonbonding in nature and can be filled with 06 electrons (Figure). The antibonding of the eg* orbital is strong, and it is not occupied at all. As a result, the number of electrons in the valence shell of these complexes would be 18 or less.
Class III:
The o splitting is the biggest in class III complexes and is relevant to good donor and acceptor ligands such as CO, PF3, olefins, and arenes at the upper end of the spectrochemical series. Because of interactions with ligand orbitals, the t2gorbital becomes bonding and should be occupied by 6 electrons. Because the eg* orbital is significantly antibonding, it is empty.
1.3.2 Electron counting
Electron counting is usually done using one of two methods:
1. Neutral atom method: For counting purposes, metal is assumed to be in a condition of zero oxidation.
2. The oxidation state of the metal is determined by evaluating the amount of anionic ligands present and the total charge of the complex.
The electron count for most compounds should add up to 18 electrons. There are, however, several exceptions to the 18 electron rule, just as there are to the octet rule.
1.3.3 Reactivity
The 18 electron rule can be used to estimate a compound's reactivity. An associative mechanism is one in which a ligand is added, whereas a dissociative mechanism is one in which a ligand is lost. A molecule will most likely undergo an associative reaction if its electron count is fewer than 18. Consider the following scenario: Would it be more likely to lose a C2H4 or gain a CO if (C2H4)PdCl2 had a 16 electron count? A 14 electron complex is formed when a C2H4 is removed, while an 18 electron complex is formed when a CO is added. We can predict that the molecule will more than likely experience an associative addition of CO based on the 18 electron rule.
1.3.4 Ligand Contributions
A list of common organometallic ligands and their electron contributions is provided below.
Neutral 2e donors | Anionic 2e donors | Anionic 4e donors | Anionic 6e donors |
PR3 (phosphines) | X- (halide) | C3H5- (allyl) | Cp- (cyclopentadienyl) |
CO (carbonyl) | CH3- (methyl) | O2- (oxide) | O2- (oxide) |
Alkenes | CR3- (alkyl) | S2- (sulfide) |
|
Alkynes | Ph- (phenyl) | NR2- (imide) |
|
Nitriles | H- (hydride) | CR22- (alkylidene) |
|
| RnE- (silyl, germyl, alkoxo, amido etc.) | OR- (alkoxide, bridging ligand) |
|
|
| SR- (thiolate, bridging ligand) |
|
|
| NR2- (inorganic amide, bridging ligand) |
|
|
| PR2- (phosphide, bridging ligand) |
|
1.3.5 Exceptions
The early transition metals (groups 3–5) have an electron count of 16 or less in general. Middle transition metals (groups 6–8) have an average electron count of 18 while late transition metals (groups 9–11) have an average electron count of 16 or lower. A structure is deemed electron-deficient or coordinately unsaturated if it has less than 18 electrons. This indicates that the compound's valence orbitals are vacant, making it electrophilic and highly reactive. When a structure has "too many electrons," it signifies that not all of the bonds are covalent, and hence some must be ionic. When compared to covalent bonds, these bonds are weaker. These organometallic compounds with an electron count larger than 18 are, nonetheless, uncommon.
1.4.1 Electron Counting and the 18 Electron Rule
The 18 Electron Rule is a valuable method for predicting organometallic complex structure and reactivity. In a simplified logic, it represents the central metal's inclination to achieve the noble gas configuration in its valence shell, and is somewhat akin to the octet rule. Depending on the energy and type of atomic and molecular orbitals, there are exceptions to this norm. 1st
1.4.2 The 18 Electron Rule
The General Rule
All of an atom's valence orbitals are usually occupied by paired electrons. The valence orbitals of transition metals are composed of ns, 3 np, and 5 (n-1)d orbitals, resulting in a tendency to be surrounded by 18 electrons. In a simplified explanation, this is similar to the octet and Lewis structural principles of major group elements.
Electron-precise structures are those that satisfy this chosen electron structure. There will be no more unoccupied low-lying orbitals accessible for further ligand coordination in transition metal complexes with 18 electrons, which are also known as saturated. Unsaturated complexes have less than 18 electron counts and can electronically bind to additional ligands.
Exceptions to The Rule
In metal complexes with strong field ligands that are good donors and acceptors, the 18 electron rule is frequently followed (for example, CO ligands). Because the energy difference (0) between the t2g and eg* orbitals is so enormous, the three t2g orbitals become bonding and are always full, whereas the two eg* orbitals are strongly antibonding and are always empty in this situation.
The antibonding feature of eg* orbitals weakens when 0 between t2g and eg* orbitals is small, as in the case of first row transition metals with weaker field ligands, and the complex can have up to 22 electrons.
In complexes of 4th and 5th row transition metals with high oxidation states, however, less than 18 electrons can be found. Because of the higher repulsion between the d orbitals of metals and the ligands, 0 is relatively significant in this scenario. The eg* orbitals are highly antibonding and remain vacant, but the t2g orbitals are non-bonding and can have 0-6 electrons occupying them.
Still, the sorts of ligands in a complex determine whether or not the complex follows the 18 electron rule.
Among the most common exceptions to the 18 electron rules are:
16-electron complexes have a low-spin metal centre that is arranged in a d8 configuration. Rh(I), Ni(II), Pd(II), and Pt(II) complexes, for example, have a square planar structure. Organometallic catalysts convert back and forth between 18 and 16 electron configurations in many catalytic processes, completing a catalytic cycle.
Bulky ligands may make the 18 electron rule more difficult to complete. Complexes with agostic interaction are an example.
The 18 electron rule is frequently broken in complexes containing ligands with strong -donating characteristics. F-, O2-, RO-, and RN2- are examples of this type of ligand.
1.4.3 Electron Counting Methods
The covalent technique and the ionic ligand approach are two extensively used methods for counting electrons in complexes. Both approaches should yield the same electron count and are applicable to any organometallic compounds.
Covalent Method
All metal-ligand bonds are deemed covalent in this manner. Ligands have no charge and can give either two, one, or zero electrons to the bond. For instance, ligands with a filled valence, such as CO and NH3, contribute two electrons. In their neutral state, halide and hydroxo groups, on the other hand, do not have an octet structure and contribute one electron to bonding. Ligands like BF3 have no free electrons, thus the two electrons needed for bonding must come from the metal centre.
The steps for using the covalent counting method are as follows:
1. Determine the metal center's group number.
2. Determine how many electrons the ligands contribute.
3. Determine the metal-ligand complex's total charge.
4. When a metal-metal bond exists, one electron is assigned to each metal centre in the bond.
5. To get the final electron count, add the group number of the metal centre and the e- count of the ligands, then factor in the overall charge of the complex.
Ionic Method
The ligands are always assigned filled valences in the ionic technique. For example, the H group, as well as other groups like halide, hydroxyl, and methyl, are now termed H-. These groups now contribute one more electron than in the covalent approach, and when a bond is formed, they oxidise the metal core. CO and NH3, for example, have a neutral charge in their octet structure and react similarly to valence techniques.
The following are the steps in the ionic counting method:
1. Determine the metal complex's total charge.
2. Determine the ligands' charges and the quantity of e-s they give.
3. Determine the amount of valence electrons in the metal centre so that the overall charge of the complex is balanced by the metal's oxidation state and the charges of the ligands. (E-count of metal centre = Metal atom group number + (charges of ionic ligands) – complex overall charge)
4. If a metal-metal bond exists, one bond equals one electron per metal atom.
5. Add the metal center's and ligands' electron counts together.
Electron Counts of Some Common Ligands
Ligand | Covalent | Ionic | Charge |
H | 1 | 2 (H-) | -1 |
Cl, Br, I | 1 | 2 (X-) | -1 |
OH, OR | 1 | 2 (OH-,OR-) | -1 |
CN | 1 | 2 (CN-) | -1 |
CH3, CR3 | 1 | 2 (CH3-,CR3-) | -1 |
NO (bent M-N-O) | 1 | 2 (NO-) | -1 |
NO (linear M-N-O) | 3 | 2 (NO+) | +1 |
CO, PR3 | 2 | 2 | 0 |
NH3, H2O | 2 | 2 | 0 |
=CRR' (carbene) | 2 | 2 | 0 |
H2C=CH2 (ethylene) | 2 | 2 | 0 |
CNR | 2 | 2 | 0 |
=O, =S | 2 | 4 (O2-, S2-) | -2 |
η3-C3H5 (π-allyl) | 3 | 2 (C3H5+) | +1 |
≡CR (carbyne) | 3 | 3 | 0 |
≡N | 3 | 6 (N3-) | -3 |
En (Ethylenediamine) | 4 | 4 | 0 |
Bipy (Bipyridine) | 4 | 4 | 0 |
Butadiene | 4 | 4 | 0 |
η5-C5H5 (cyclopentadienyl) | 5 | 6 (C5H5-) | -1 |
η6-C6H6 (benzene) | 6 | 6 | 0 |
η7-C7H7 (cycloheptatrienyl) | 7 | 6 (C7H7+) | +1 |
Examples
Examples of Electron Counting of Some Organometallic Complexes | |||
Complexes | Covalent Method | Ionic Method | Total Number of Electrons |
(η5-C5H5)2Fe | ● Fe gives 8e- ● 2 η5-C5H5 give 2×5e- ● Complex Charge is 0 | ● Fe(II) gives 6e- ● 2 η5-C5H5- give 2×6e- | 18 |
[V(CO)7]+ | ● V gives 5e- ● 7 CO give 7×2e- ● Complex Charge is +1 (-1e-) | ● V(I) gives 4e- ● 7 CO give 7×2e- | 18 |
[Re(CO)5(PF3)]+ | ● Re gives 7e- ● 5 CO give 5×2e- ● PF3 gives 2e- ● Complex Charge is +1 (-1e-) | ● Re(I) gives 6e- ● 2 CO give 5×2e- ● PF3 gives 2e- | 18 |
1.4.4 Electron donation of common ligands
Some common transition metal ligands and the number of electrons they contribute to a metal centre are included in the table below. Some ligands have the ability to contribute a different quantity of electrons. For example, depending on the hybridization of the oxygen atom, an alkoxide, M-OR, can give two to six electrons.
1.5.1 Organometallic Chemistry of d Block Metals
Transition metal organometallic chemistry is a relatively new field. Although Zeise's salt (K[PtCl3(C2H4)], tetracarbonylnickel (Ni(CO)4), and pentacarbonyliron (Fe(CO)5), all of which are now categorised as organometallic compounds, were synthesised in the 19th century, their bonding and structures remained unknown. W. Hieber's and others' work on metal carbonyl compounds was crucial in the 1930s, but the results were limited due to the immature structural analysis techniques available at the time.
The discovery of ferrocene, Fe(C5H5)2, in 1951 was a watershed moment in this field's chemistry. Single crystal X-ray structural analysis, NMR spectra, infrared spectra, and other techniques revealed the complex's very unique bonding behaviour, which provided as a springboard for further research in the field. Despite the widespread belief that transition metal-carbon bonds are highly unstable, the finding of ferrocene's exceptional thermal stability was a huge breakthrough. It was also shown that the molecule had a sandwich structure, with the five carbon atoms of the cyclopentadienyl groups bonding to the core metal iron at the same time. While various coordination modes of hydrocarbon ligands were discovered one after the other, the industrial importance of organometallic transition metal compounds grew with the discovery of olefin polymerization catalysts (Ziegler catalyst), homogeneous hydrogenation catalysts (Wilkinson catalyst), and development of asymmetric synthesis catalysts, among other things. This significance was recognised by the Nobel Prizes given to K. Ziegler, G. Natta (1963), E. O. Fischer, and G. Wilkinson (1973).
According to the definition of an organometallic compound, there must be at least one direct bond between a metal and a carbon atom, however CN complexes and other compounds with no organometallic property are frequently eliminated. Metal carbonyl compounds are organometallic in many ways, including bonding, structure, and reactions, and thus are an excellent model system for learning about transition metal organometallic chemistry.
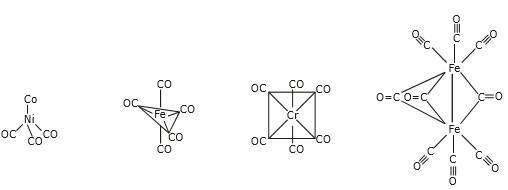
(a) Metal carbonyl compounds
Binary metal carbonyl compounds containing only a metal and CO ligands are commonly made by reacting a highly reactive metal powder with carbon monoxide directly, or by reducing a metal salt to zero valence and then reacting with high-pressure carbon monoxide. Tetracarbonylnickel, on the other hand, is formed by the interaction of nickel metal with carbon monoxide at room temperature and atmospheric pressure. It was originally found near the end of the nineteenth century. Other metal carbonyl compounds, on the other hand, necessitate high temperatures and pressures for synthesis.
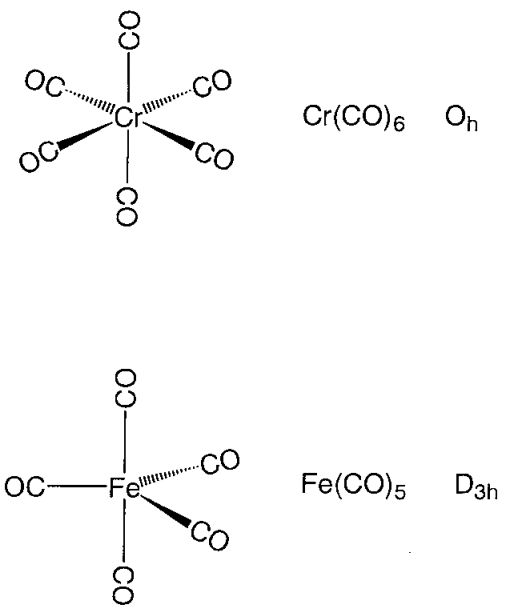
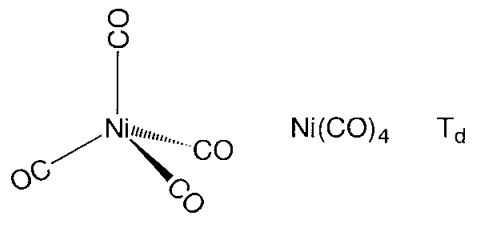
Figure: - Structures of metal carbonyl compounds.
The coordination structures of mononuclear metal carbonyl compounds are highly symmetric polyhedral. Hexa-coordinate chromium, molybdenum, and tungsten hexacarbonyl, M(CO)6, has a regular octahedral coordination structure, while penta-coordinate pentacarbonyliron, Fe(CO)5, has a triangular bipyramid coordination structure, and tetracarbonylnickel, Ni(CO)4, has a regular tetrahedron coordination structure. Carbonyl ligands have carbon atoms that are aligned with the metal, and CO moieties are positioned along the metal-carbon axis. Mn2(CO)10 is a binuclear metal carbonyl with a Mn-Mn link connecting two square pyramidal Mn(CO)5 components. Two Fe(CO)3 subunits are joined by three CO ligands in Fe2(CO)9, and two Co(CO)3 subunits are coupled by three CO bridges and a Co-Co link in Co2(CO)8.
There exist several cluster metal carbonyl compounds with metal-metal bonds connecting three or more metals, and terminal CO, -CO (a bridge between two metals), and 33-CO (a bridge capping three metals) are all coordinated to the metal frames. A pyrolysis reaction of mononuclear or binuclear carbonyl compounds produces a large number of cluster carbonyls. Table lists common metal carbonyl compounds and their characteristics.
| 5 | 6 | 7 | 8 | 9 | 10 |
4 | V(CO)6 | Cr(CO)6 | Mn2(CO)10 | Fe(CO)5 | Co2(CO)8 | Ni(CO)4 |
5 |
| Mo(CO)6 | Tc2(CO)10 | Ru3(CO)12 | Rh6(CO)16 |
|
6 |
| W(CO)6 | Re2(CO)10 | Os3(CO)12 | Ir4(CO)12 |
|
1.5.2 Back donation
Carbon monoxide is bonded to a zero-valent metal to form a metal carbonyl complex. It had been unknown for a long time why such bonding was even possible, let alone stable. The coordination hypothesis of A. Werner was founded on the notion that normal coordination bonds were created by the donation of electrons from highly basic ligands to a metal. Because carbon monoxide has a low basicity and transition metal-carbon bonds are not particularly stable, a plausible explanation for the stability of metal carbonyl compounds was sought. A bonding interaction between the metal and carbon is expected if the shape and symmetry of the metal d orbital and the CO (antibonding) orbital for the carbon-oxygen bond are adequate for overlap. From this standpoint, the bonding method depicted in Figure was developed. Back donation is the process by which electrons from the loaded metal d orbital are donated to the unoccupied carbon monoxide orbital. Back-donation stabilises the M-C bond by preventing the collection of extra electrons on a metal atom with a low oxidation state.
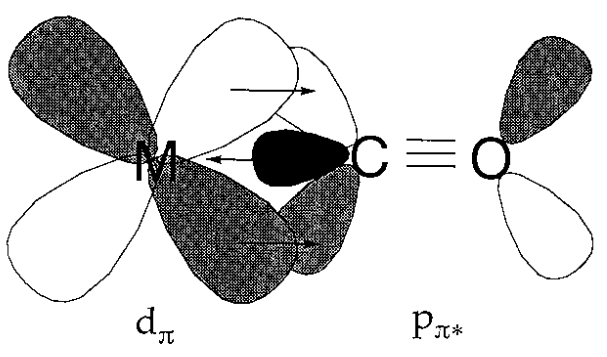
Fig- Back donation in metal carbonyls.
In vibrational spectra, an increase in the order of the metal-carbon bond is indicated by an increase in the M-C and a decrease in the C-O stretching frequencies. Carbonyl frequencies are easily observable, hence infrared spectra are relevant. The passage of negative charge from a metal's coordinated ligands lowers its oxidation state, which results in a drop in C-O stretching frequencies.
(b) Hydrocarbon complexes
A metal-carbon bond exists in an organometallic complex, and between one and eight carbon atoms in a hydrocarbon ligand bond to a metal. The number of atoms in a ligand that have direct coordinative interaction with the metal is called hapticity, and it is added to. 55% (pentahapto)-cyclopentadienyl is an example.
A radical is a ligand that provides an odd number of electrons to a metal and is stabilised by attaching to it. A neutral molecule is one that provides an even amount of electrons to a metal and is stable even if it is not attached to the metal. Exceptions to this norm are carbene or carbyne ligands. In many circumstances, the chemical formula of an organometallic compound is written without the square brackets [ ] that are typical of such a complex, and we will adopt this tradition in this text.
Table: Hapticity and number of donating electrons of hydrocarbon ligands | |||
Name | Hapticity | Number of electrons | Example |
Alkyl | η1η1 | 1 | W(CH3)6 |
Alkylidene | η1η1 | 2 | Cr(CO)5{C(OCH3)C6H5} |
Alkene | η2η2 | 2 | K[PtCl3(C2H4)] |
ππ-allyl | η3η3 | 3 | Ni(η3η3-C3H5)2 |
Diene | η4η4 | 4 | Fe(CO)3(η4η4-C4H6) |
Cyclopentadienyl | η5η5 | 5 | Fe(η5η5-C5H5)2 |
Arene | η6η6 | 6 | Cr(η6η6-C6H6)2 |
Tropylium | η7η7 | 7 | V(CO)3(η8η8 -C7H7) |
Cyclooctatetraene | η8η8 | 8 | U(η8η8 -C8H8)2 |
1.5.3 Alkyl ligands
M-C single bonds are found in alkyl or aryl transition metal complexes. Despite numerous attempts over the course of chemical history, they were never successfully isolated, and it was long assumed that all M-C bonds were inherently unstable. Only in the 1950s did stable alkyl complexes begin to be created. Some representative compounds include Cp2ZrCl(Pr),WMe6, CpFeMe(CO)2, CoMe(py)(dmg)2, IrCl(X)(Et)(CO)(PPh3)2, NiEt2(bipy), and PtCl(Et)(PEt3)2. The reactions of compounds having M-halogen bonds with main-group metal-alkyl compounds, such as a Grignard reagent or an organolithium compound, are frequent synthetic routes among the numerous synthetic procedures so far established. Vitamin B12, whose structure was established by D. Hodgkin (1964 Nobel Prize), is known to have a particularly stable Co-C bond. Homoleptic alkyls, such as WMe6, are metal alkyl compounds with only an alkyl ligand.
The low activation energy of alkyl complexes' breakdown, rather than a low M-C bond energy, is rapidly becoming regarded as a primary reason of their instability. Elimination is the most common method of breakdown. A hydrocarbon ligand's bonding interaction with the core transition metal results in the creation of a metal hydride and an olefin. An agostic interaction is a type of interaction like this. Although alkyl and aryl ligands are one-electron ligands, they are considered anions when the metal's oxidation number is calculated. In this way, the hydride ligand, H, is similar to the alkyl ligand.
1.5.4 π allyl complexes
An allyl group, CH2=CH-CH2-, is a 1-electron ligand like an alkyl group when it is connected to a metal via a carbon atom. Three carbon atoms link to the metal concurrently as a 3-electron ligand if the double bond delocalizes. This is an odd electron ligand that is nominally anionic and is stabilised by being coordinated to the metal.
Examples include Pd(C3H5)(Ac)(PPh3), Co(C3H5)3, and others. Various reactions occur in the catalytic reactions of unsaturated hydrocarbons because coordination modes of 11, 22, and 33 are feasible.
Complexes of 1.5.5 cyclopentadienl
C5H5 is the abbreviation for the cyclopentadie nyl ligand. C5Me5, a beneficial ligand dubbed Cp star and denoted by Cp*, is a helpful ligand in which the hydrogen atoms of Cp are substituted by methyl groups. Ferrocene (Cp2Fe) is an orange-colored iron compound with two cyclopentadienyl groups attached to it. It was found in two laboratories separately, although the discoverers postulated incorrect structures. G. Wilkinson's group, which won a Nobel Prize, clarified the right structure (1973). The following chemical route is commonly followed in the synthesis of ferrocene:
2C5H6+2Na→2Na(C5H5)+H2 (6.4.1)
FeCl2+2Na(C5H5)→Fe(C5H5)2+2NaCl (6.4.2)
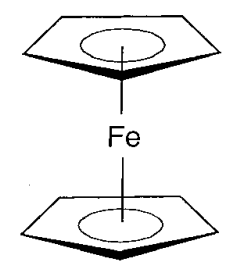
Figure - Structure of ferrocene.
The structure of ferrocene is an iron atom wedged between two C5H5 rings, according to single crystal X-ray structural study (Figure). In ferrocene, five carbon atoms link to the iron at the same time, and unsaturated C-C bonds delocalize in the five-membered rings. Because this type of connection had never been seen before, it piqued people's interest, and several derivative compounds were created, as well as a wide range of chemistry.
| 4 | 5 | 6 | 7 | 8 | 9 | 10 |
4 | Cp2TiCl2 | Cp2V | Cp2Cr | Cp2Mn | Cp2Fe | Cp2Co | Cp2Ni |
5 | Cp2ZrCl2 | Cp2NbCl2 | Cp2MoCl2 | Cp2TcH | Cp2Ru |
|
|
6 | Cp2HfCl2 | Cp2TaCl2 | Cp2WCl2 | Cp2ReH | Cp2Os |
|
|
The cyclopentadienyl ligand is a formally anionic 5-electron ligand. A 1-electron ligand, like an alkyl group, is formed when only one of the five carbon atoms is linked to a metal. In rare situations, it becomes a 3-electron ligand and coordinates to a metal as a -allyl system that spans three carbon atoms. The reactivity of the Cp group of ferrocene is similar to that of aromatic compounds. Because the Cp group has played such an important role as a stabilising ligand in realising the synthesis of novel compounds with new metal-ligand bonding modes, it is reasonable to conclude that it has contributed more to organometallic chemistry than any other ligand. Although ferrocene has two parallel Cp rings connected to the metal, Cp2TiCl2 and Cp2MoH2 contain bent Cp ligands and are referred to as bent-sandwich compounds.
1.5.6 Olefin complexes
The oldest known organometallic compound is Zeise's salt, K[PtCl3(C2H4)], which was produced and studied by Zeise about 1825, however its coordination structure was only hypothesised in 1954 and confirmed by neutron diffraction in 1975. The Dewar-Chatt-Duncanson model describes the manner of coordination of an olefin to a transition metal, and the bond between the metal and the olefin is stabilised by the contribution of d-p back donation. An olefin is a two-electron ligand, and many olefin complexes have a core metal that is in a low oxidation state. Dienes or trienes with two or more double bonds act as 4-electron or 6-electron ligands to bind to metals. Examples are Fe(CO)3(C4H6) and Ni(cod)2, in which a butadiene or cyclooctadienes (cod) are coordinated to the metal. Because cyclooctadienes are easily removed from Ni(cod)2, it's an easy way to get atomic, zero-valent nickel. This compound is also known as "bare nickel."
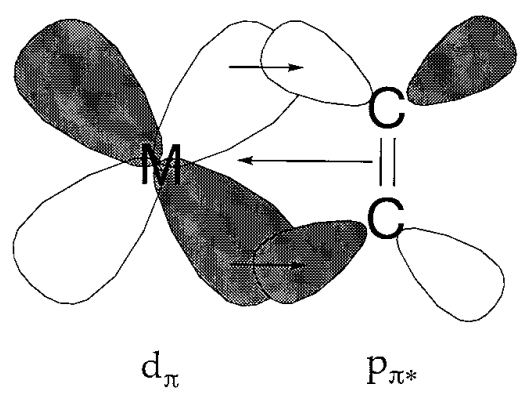
Figure- Back-donation in olefin complexes.
1.5.7 Arene complexes
Aromatic compounds are 6-electron donors with six carbon atoms that coordinate to transition metals in the 66 coordination mode. Cr(C6H6)2 bisbenzenechromium is an example of such a chemical. It's made by reducing chromium chloride in benzene and features a sandwich structure with a chromium atom sandwiched between two benzene rings. Cr(CO)3(C6H6) is formed when a benzene ligand is substituted by three carbonyls.
1.5.8 18 electron rule
The importance of counting valence electrons in chemistry cannot be overstated. Changes in the quantity of valence electrons have a significant impact on a compound's bonding, structure, and reactions. Because organometallic compounds contain both metal and organic components, calculating the amount of electrons becomes difficult. Hydrocarbyl ligands are classed as neutral molecules that coordinate to the metal or radicals that bond to the metal, with radicals such alkyls and cyclopentadienyl being dubbed anionic ligands. The radical ligand becomes formally anionic when one electron from the metal is transferred to it. When counting the number of valence electrons, it's easier to remember that both the metal and the ligands are neutral. Table lists the number of donor electrons in typical carbon ligands from this perspective. The amount of donor electrons supplied by the ligand varies depending on the number of ligating atoms that have coordinative interactions with the metal, even within the same ligand. Depending on the nature of coordinative interactions with the metal, a cyclopentadienyl ligand can contribute 1, 3, or 5 electrons.
A transition metal organometallic complex has good thermal stability when the total number of valence electrons of the metal and ligands is 18. The 18 electron rule is satisfied by Cr(CO)6, Fe(CO)5, Ni(CO)4, Fe(C5H5)2, Mo(C6H6)(CO)3, and so on, but the monomeric components of Mn2(CO)10, Co2(CO)8, or [Fe(C5H5)(CO)2]2 have only 17 electrons, and the extra electron comes from the companion metal via a metal-metal connection. The 18 electron rule, unlike the 8 electron rule in main group chemicals, has a limited application. That is to say, it is a sufficient condition, but high thermal stability compounds are not always 18 electron compounds.
Although many organometallic compounds with carbonyl or cyclopentadienyl ligands from Group 6 (chromium group) to Group 9 (cobalt group) satisfy the 18 electron rule, many compounds from the early transition metals (Group 3 - 5) and Group 10 (nickel group) do not. The 18 electron rule is not satisfied by W(CH3)6 (12e), TiCl2(C5H5)2 (16e), and IrCl2(CO)(PPh3)2 (16e), V(CO)6 (17e), Co(C5H5)2 (19e), Ni(C5H5)2 (20e), and other compounds. The 18 electron rule, on the other hand, can help you figure out which bonding modes are present in a given complex. For example, Fe(C5H5)2(CO)2 with two pentahapto cyclopentadienyl ligands has 22 electrons formally, while the molecule has 18 electrons if one of the ligands is monohapto. This is the actual coordination of this complex, according to structural analysis.
(c) Phosphine complexes
PX3 tertiary phosphines are excellent stabilising ligands for transition metal complexes, and they coordinate to metals in a wide range of oxidation states. In the chemistry of organometallic complexes, phosphones are widely utilised as carbonyl or cyclopentadienyl ligands. PX3 are Lewis bases that coordinate to metals utilising the lone pair on phosphorus and exhibit -acidity when substituents X, such as Ph, Cl, or F, have significant electron accepting characteristics. PX3's electrical versatility is what allows it to generate so many compounds. In general, the -acidity decreases in the following order: PF3 > PCl3 > PPh3 > PR3. Typical substituted phosphines are triphenylphosphine and triethylphosphine. Table lists the tertiaryphosphine complexes, which are mostly metal halide complexes. Only a few phosphine complexes are formed by manganese, Mn, and the early transition metals.
Table: Typical tertiary phosphine complexes (dmpe = 1,2-bisdimethylphosphinoethane; dppe = 1,2-bisdiphenylphosphinoethane) | ||||||||
| 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
4 | [TiCl4(PPh3)2] | [VCl3(PMe | [CrCl2 | [Mn(CO)4 | [FeCl2 | [CoCl2 | [NiCl2(PEt3)2] | [CuBr(PEt3)]4 |
5 | [ZrCl4(dppe)] | [NbCl4(PEt | [MoCl3(PMe | [TcCl3(PMe2 | [RuCl2 | [RhCl(PPh3)3] | [PdCl2(P | [AgCl(PPh3)] |
6 | [HfCl4(dppe)] | [TaCl4(PEt3)2] | [WCl4(PPh3)2] | [ReCl3(PMe2 | [OsCl3 | [IrCl3(PPh3)3] | [PtCl2(P | [AuCl(PPh3)] |
By changing the halogens in the phosphine complexes, a variety of derivatives can be made. Polydentate phosphine complexes with more than two coordination sites, as well as monodentate phosphine complexes, have been created, and they are also used as stabilising ligands in hydride, alkyl, dinitrogen, and dihydrogen complexes. The coordination of optically active phosphines in rhodium or ruthenium complexes makes them good catalysts for asymmetric synthesis.
1.6.1 Carbon Monoxide and backbonding:
Carbon monoxide is a straightforward but intriguing ligand. Carbon monoxide, although being a poor base, is a powerful field ligand due to the presence of backbonding, as we previously stated.
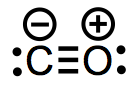
General Properties
CO is a dative, L-type ligand that increases the overall electron count by two units without changing the oxidation state of the metal centre. We've recently discovered that the carbonyl ligand has two bonding interactions: a ligand-to-metal n d interaction and a metal-to-ligand d* interaction. Because the metal donates electron density back to the ligand, this contact is known as backbonding. I like to use the right-hand resonance structure whenever feasible to remind myself of the existence of backbonding; but, no matter what resonance form is drawn, CO must be treated as an L-type ligand.
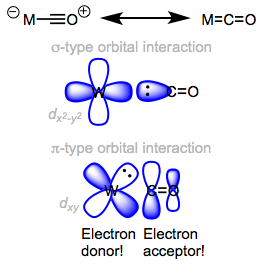
Orbital interactions in M=C=O.
CO is a good -acceptor (or -acid) and a strong -donor (or -base). The identification of the metal centre has a significant impact on the characteristics of ligated CO. The relevance of backbonding in metal carbonyl complexes is dictated by the electronic characteristics of the metal centre. To put it clearly, metal centres with more electrons are better at backbonding to CO.
The use of infrared spectroscopy to objectively support the concept of backbonding is well-known. The frequency corresponding to the C=O stretching mode is given in the table below, which organises several metal carbonyl complexes in “periodic” order. It's worth noting that every complexed CO has a stretching frequency that is lower than that of free CO. The culprit is backbonding! Complexed carbon monoxide has a lower C–O bond order than free CO (nearly always).
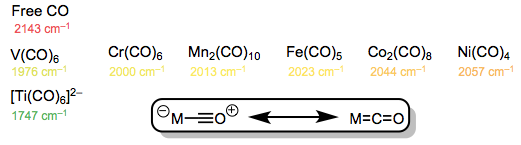
C=O stretching frequencies in metal-carbonyl complexes. Does something seem off here?
As we proceed left to right through the periodic table, we see a definite rise in frequency (an increase in C–O bond order). When we consider that the amount of d electrons in the neutral metal rises as we move left to right, this discovery may appear strange. Shouldn't metal centres with more d electrons (and hence be more "electron rich") be better at backbonding? What exactly is going on here? Remember the orbital energy's periodic trend? The d orbital energy fall as we move left to right, and the energies of the d and * orbitals separate. As a result, as we progress toward the more electronegative late transition metals, the backbonding orbital interaction grows worse (remember that strong orbital interactions require well-matched orbital energies)! Enamines and enol ethers are analogous to enamines and enol ethers in organic chemistry. In enol ethers, the more electronegative oxygen atom is a worse electron donor than the nitrogen atom in enamine.
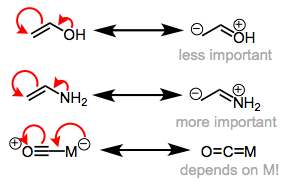
The relevance of backbonding is determined by the metal's electronegativity and electron density.
Other ligands on the metal core also play a role in backbonding, which should not be overlooked. Backbonding should be strengthened by electron-donating ligands (they make the metal a greater electron donor), while backbonding should be weakened by electron-withdrawing ligands. Due to better backbonding, adding electron-rich phosphine ligands to a metal core, for example, reduces CO stretching frequency.
Carbonyl ligands are well-known for their ability to connect numerous metal centres. Depending on the structure of the complex and the bridging mode, bonding in bridged carbonyl complexes can be either “traditional” or “delocalized.” The different electron donors and acceptors present on the CO ligand result in a wide range of bridging modes (and the possibility of delocalized bonding). The figure below depicts known bridging modes.
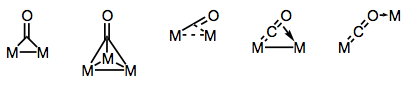
Building bridges with carbonyl ligands!
1.6.2 Pi Donor and Acceptor Ligands
Along with other qualities such as metal identity and oxidation state, the type of ligands bound to the central metal is a significant property of a complex molecule. The molecular orbitals are determined by the identity of the ligand and, as a result, the ability of the ligand to donate or accept electrons to the centre atom.
The progression of substances as weak field to strong field ligands can be seen in the spectrochemical series. The -bonding interactions of ligands can also be used to classify them. This interaction indicates the amount of energy split between the eg and t2g energy levels of the molecular orbitals, which determines the ligands' field strength.
X-, OH-, and H2O are examples of weak field ligands; H-, NH3, CO, and PR3 are examples of strong field ligands.
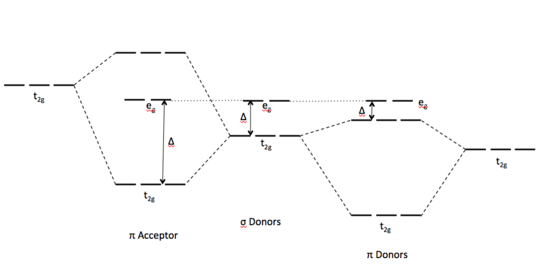
Figure: MO Diagrams of Pi Donor Ligands and Pi Acceptor Ligands.
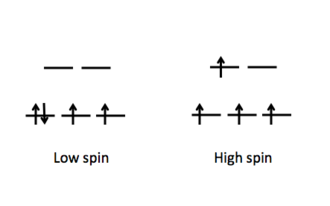
Figure: Electron configuration of high and low spin.
Electron configuration of high and low spin.
The SALCs of the ligands are occupied in a -donor ligand, therefore it transfers electrons to the molecular * and * orbitals. Because the orbitals linked with eg are not involved in interactions, it maintains its energy level (Figure). The occupied ligand SALC t2g orbitals (ie. Dxy, dxz, dyz) that would form molecular orbitals with the metal t2g orbitals (ie. Dxy, dxz, dyz) have a lower energy than their metal counterparts. The resulting MO has * orbitals that are less energetic than the * orbitals generated by the non-bonding orbitals (eg). , split denotes the difference between the t2g* and eg orbitals. The is tiny in the -donor instance due to the low * level.
Because the t2g SALCs of pi accepting orbitals are vacant, they have a higher energy than metal t2g orbitals. The t2g* orbitals that result are higher than the * orbitals. This results in a wider gap between the eg and t2g orbitals, resulting in high split ligands for these -accepting orbitals.
Finally, the size of as determined by the ligand's identity will determine how electrons are dispersed in metal d orbitals. Weak field ligands cause a small spin configuration, resulting in a high spin configuration. Strong field ligands induce a large spin configuration on the d electrons, resulting in a low spin configuration.
1.6.3 How to rationalise with MO theory that CO is a two-electron donor through carbon?
The main distinction between these molecules is that CO is heteronuclear, which means that the energy levels of the molecular orbitals and the atoms will differ.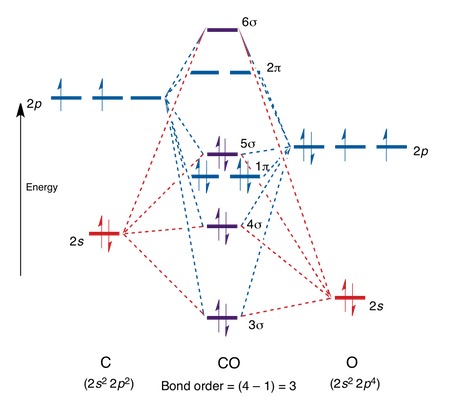
The HO is represented by the number 5 in the MO diagram above.
1.7.1 General Information
Carbonyl Complexes are ligand-coordinated complexes that include carbon monoxide. Carbon monoxide is a frequent ligand in transition metal chemistry, owing to its synergistic bonding properties with transition metals. The bonding of CO to a metal can be described as having two components. The first component is a two-electron donation of the lone pair on carbon into a vacant metal d-orbital (coordination solely through oxygen is extremely unusual). The metal becomes more electron rich as a result of the electron donation, and to compensate for this higher electron density, a filled metal d-orbital may interact with the empty pi* orbital on the carbonyl ligand to relieve itself of the extra electron density. Pi-backbonding or pi-backdonation is the name for the second component. This is depicted both graphically and through a simple MO image below. Each component of the bonding interaction has been colour coloured on the MO for easy identification:
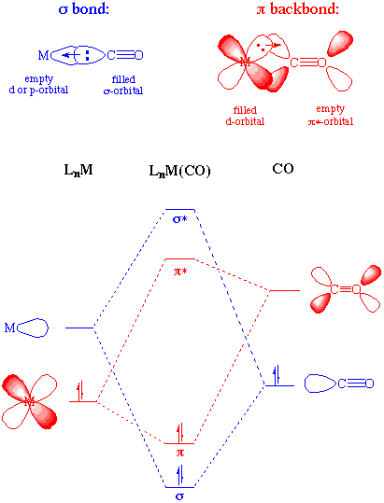
The two elements of this relationship are mutually beneficial. The stronger the pi-backbonding connection, the greater sigma donation by the carbonyl (or other sigma-donors on the metal centre). It's worth noting that, despite the possession of a pi* orbital on the CO, this is still a bonding interaction in terms of the metal centre. The nature of carbonyl-metal bonding is fundamentally comparable to that of alkenes, acetylenes, phosphines, and dihydrogen.
The occupancy of the pi* on CO does result in a decrease in bond order within the carbon monoxide molecule. As the pi-backdonation becomes stronger, the CO bond order should fall below that of the free ligand, as expected. The lengthening of the C-O bond and a decrease in the carbonyl stretching frequency in the IR are two possible outcomes if the CO bond order is lowered. Both of these statements are correct.
1.7.2 Bonding modes of CO
CO usually attaches to carbon in an end-on fashion, as shown above. Bridging carbonyls, on the other hand, are common and frequently undergo exchange with terminal carbonyls. The "semi-bridging" carbonyl is a type of bridging carbonyl with an asymmetric rather than symmetric M-CO-M link. The metrical data provided in each case is consistent with a decrease in the CO bond order: Some less prevalent bonding modes are shown below; observe that the metrical data provided in each case is consistent with a drop in the CO bond order:
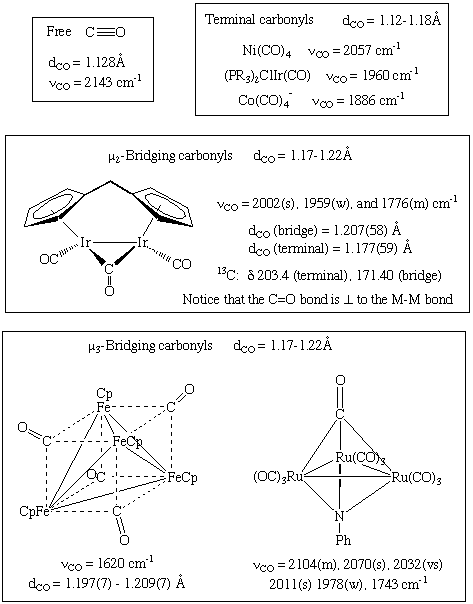
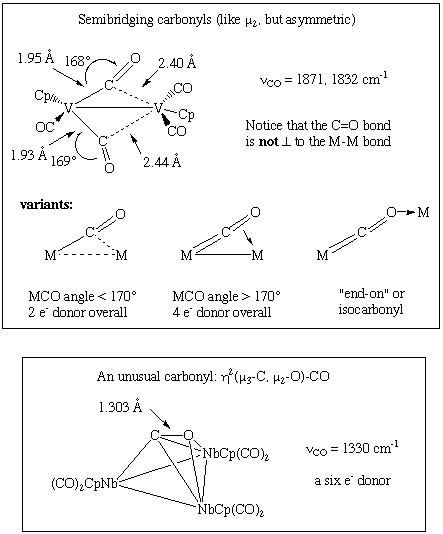
1.7.3 Spectroscopic Features of Carbonyl Complexes
Stretching frequencies in the infrared range from 2125 to 1850 cm-1 for uncoordinated or "free" CO and 2125 to 1850 cm-1 for terminal M-CO.
1850 to 1750 cm-1 Doubly bridging (mu-2)
1675 to 1600 cm-1 for triple bridging (mu-3)
Semibridging is a phrase that refers to a state that is halfway between terminal and mu-2.
The IR spectra of carbonyl compounds show two useful features that are both consistent with the concept of pi-backbonding mentioned above:
The CO stretching frequency drops by around 100 cm-1 with each charge applied to the metal centre
The lower the CO stretching frequency, the better the sigma-donating capability (or the worse the pi-acceptor capability) of the other ligands on the metal.
Counting the number of IR and Raman CO stretching frequencies allows one to make a structural assignment for simple carbonyl compounds. Group theory can be used to predict the number of CO stretches expected for certain geometries/isomers, and the computed findings can then be compared to experimental data.
Coordinated carbonyl ligands often appear in the range of 180 to 250 ppm in the 13C NMR spectra. Carbonyl compounds with isotopically enriched isotopes are frequently synthesised to simplify mechanistic research or to make spectrum gathering easier. Furthermore, connecting a 13C-enriched compound to additional spin active nuclei like 103Rh or 31P can help with structural assignments.
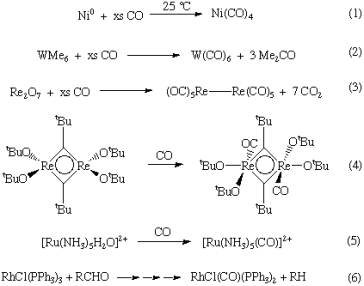
1.7.4 Syntheses of metal carbonyls
Metal carbonyls can be created in a number of different methods. Here are a couple such examples:
Direct interaction with the metal can produce homoleptic or binary metal carbonyls for Ni and Fe (Equation 1).
In other circumstances, a metal precursor is reduced in the presence of CO (or with CO as the reductant) (Equations 2-3).
Carbon monoxide interacts with a variety of metal complexes, most commonly occupying a vacant coordination site (Equation 4) or performing ligand substitution events (Equation 5). (Equation 5).
CO ligands are occasionally produced through a deinsertion event involving a coordinated ligand (Equation 6).
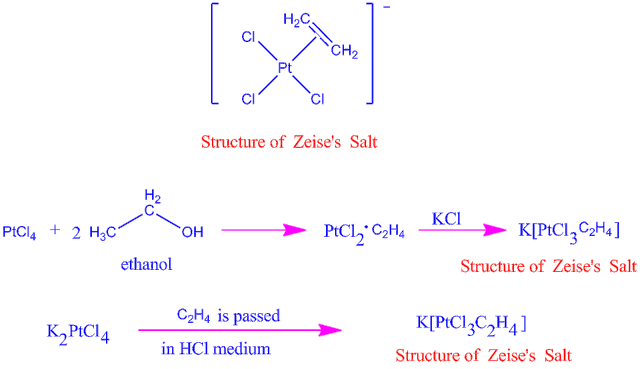
1.8.1 What is Zeise’s salt? Discuss the structure and bonding of Zeise’s salt?
Platinum (Pt), a 4d transition metal, which forms a large number of organo-metallic compounds. Zeise’s salt is one of them.
The salts containing the ion [C2H4PtCl3] -1 are known as Zeise’s salt after the name of discover such salts may be K[C2H4PtCl3], Na[C2H4PtCl3], NH4[C2H4PtCl3] etc.

1.8.2 Preparation of Zeise’s salt
When platinum tetra chloride reacts with ethanol, it forms an adduct, which when combined with KCl forms Zeise's salt. The two methods for making Zeise's salt are shown here.
The nature of bonding in an olefinic compound like Zeise's salt is still unknown, although it is obvious that there are no localised -bonds between metal atoms and carbon.
The interaction between the -electrons in the unsaturated molecule and the metal atom's hybrid orbitals is commonly attributed to this. The X-ray diffraction method has been used to investigate several olefin compounds.
The coordinated double bond in complexes like [C2H4PtCl2]2 and [(C2H4)2RhCl]2 is normal to the coordination plane, and the [ C2H4PtCl3] -1 ion is comparable.
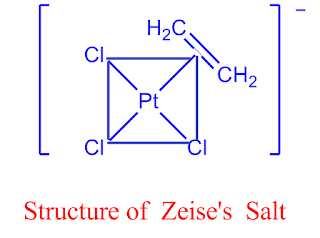
The molecular orbital (M.O) technique can explain the bonding in Zeise's salt. The metal-to-olefin bond is thought to be made up of two pieces, according to the notion.
(I) Overlap of the olefin's -electron density with a metal atom's -type acceptor orbital.

(II)Back bonding caused by electron density flow from a filled metal d orbital into antibonding unoccupied C-atom orbitals.

This perspective is related to carbonyl bonding, but differs somewhat, and is referred to as -bonding.
1.8.3 Why the C –C olefinic bond length in Zeise’s salt is greater than the C –C bond in free hydrocarbon?
Back bonding from a suitable full metal d orbital into the unoccupied anti bonding*orbital on the carbon atom occurs in Zeise's salt. The C–C bond order should be decreasing in the unbound C2H4 molecule.
As a result, the C–C bond length in Zeise's salt increases from 1.34 for a free C2H4 molecule to 1.4-1.47 for a coordinated C2H4 molecule.
1.8.4 Why the ethylene molecule is perpendicular to the PtCl3 molecular plane in Zeise’s salt?
In Zeise’s salt the metal ion, Pt(II) contains three π-type filled d-orbital which are dxy, dyz, and dxz.
When the coordinated ethylene molecule lies perpendicular to the molecular plane , the back bonding may take place either through dyz or dxz filled orbitals , but it may take place only through dxyfilled orbital.
When C2H4 molecule exists in the plane of the molecule . As a consequence, C2H4 molecule lies lies perpendicular to the PtCl3 molecular plane for better scope of back bonding.
1.8.5 Why Cl – ion trans to the ethylene has the large Pt –Cl bond distance than the other two Pt –Cl bond distance in Zeise’s salt?
The trans orienting power of ligands can be used to explain the above finding.
Due to back bonding, the metal olefin bond has multiple bond character, and this multiple bond character (-bonding effect) reduces the metal–carbon bond length.
As a result, the metal chlorine connection linked to the ethylene molecule weakens, resulting in an increase in the length of the Pt–Cl bond.
However, this -bonding has no effect on the Pt–Cl bond character cis to the C2H4 molecule.
As a result, the trans Pt–Cl bond distance is greater than the other two Pt–Cl bond distances.
1.8.6 Is the rotation of C2H4 molecule in Zeise’s salt hampering the stability of the complex?
The ethylene molecule can either lay on the same plane as the PtCl3 unit or lie perpendicular to the plane of the molecule, based on bonding in Zeise's salt.
Back bonding is conceivable in each scenario. Back bonding occurs in the first scenario via a filled dxy metal orbital, while it occurs in the second instance via dyz or dxz metal orbitals.
Because the probability of such back bonding is larger in the second situation, the stability is increased.
Experimental measurements have revealed that the energy barrier of the C2H4 molecule is only 5 – 6 kcal per mole, implying that the stability difference is on the order of 5 – 6 kcal per mole.
As a result of the previous reasoning, it is evident that in Zeise's salt, free rotation of the C2H4 molecule is conceivable.
1.9.1 Synergic effect in metal carbonyls
In enol ethers, the more electronegative oxygen atom is a worse electron donor than the nitrogen atom. Consider the metal carbonyl, which is also known as pentacarbonyl iron (0). Carbonyl ligands are well-known for their ability to connect numerous metal centres. CO is a dative, L-type ligand that increases the overall electron count by two units without changing the oxidation state of the metal centre. Depending on the structure of the complex and the bridging mode, bonding in bridged carbonyl complexes can be either “traditional” or “delocalized.” Metal carbonyl synthesis. Preparation and structure of Zeise's salt, as well as proof of synergic effect and a comparison of synergic effect with that of carbonyls. As we proceed left to right through the periodic table, we see a definite rise in frequency (an increase in C–O bond order). As a result, axial metal-ligand bonds are weaker than equatorial metal-ligand connections. The different electron donors and acceptors present on the CO ligand result in a wide range of bridging modes (and the possibility of delocalized bonding). The relevance of backbonding in metal carbonyl complexes is dictated by the electronic characteristics of the metal centre. CO is a dative, L-type ligand that increases the overall electron count by two units without changing the oxidation state of the metal centre. As a result of the metal-ligand association, a synergic effect is formed. Synergic effect refers to the unique trait of back bonding that stabilises the metal ligand interaction. Between the core metal atom, iron, and the neutral carbonyl ligands, there is synergic bonding. The frequency corresponding to the C=O stretching mode is given in the table below, which organises several metal carbonyl complexes in “periodic” order. It's a bond between a metal and a Carbonyl group serving as a ligand, Kopur. Here are a couple such examples: Direct interaction with the metal can produce homoleptic or binary metal carbonyls for Ni and Fe (Equation 1). This enhances the metal-ligand bond, but it weakens the bond because electron density from the filled orbitals of the Iron atom is pushed into the anti bonding orbital. The relevance of backbonding is determined by the metal's electronegativity and electron density. The d orbital energy fall as we move left to right, and the energies of the d and * orbitals separate. Between the core metal atom, iron, and the neutral carbonyl ligands, there is synergic bonding. As a result, as we progress toward the more electronegative late transition metals, the backbonding orbital interaction grows worse. The identification of the metal centre has a significant impact on the characteristics of ligated CO. The metal-carbon bond in a metal carbonyl possesses the properties of both and bonds.
Key takeaway:
● Gilman reagents, which contain lithium and copper, and Grignard reagents, which contain magnesium, are examples of organometallic compounds. Organometallic compounds containing transition metals include tetracarbonyl nickel and ferrocene.
● Organometallic compound classification: Organometallic compounds are divided into two categories based on the type of the metal-carbon bond: ′′ Ionic bonded organometallic compounds: Alkali, alkaline earth metals, Lanthanides, and Actinides all have organometallic compounds that are primarily ionic.
● Metal displacement, metathesis, and hydrometallation are the four M-C bond formation reactions of a metal with an organic halide that can be used to make most organometallic compounds. When the displacing metal is higher in the electrochemical series than the displaced metal, transmetallation is advantageous.
● Organometallic compounds are those in which the carbon atoms of organic groups are directly linked to metal atoms. Organometallics are made up of elements including boron, phosphorous, silicon, germanium, and antimony that have an organic group attached to them.
● The electropositive character of the metal atom or group has a significant impact on the reagent's reactivity. The most reactive of the regularly used compounds in this family are alkyllithium (and sodium) compounds, which have metal-carbon bonds that are around 30% ionic.
References:
1. Das Asim K., Fundamentals of Inorganic Chemistry, Vol. II, CBS Publications, 2nd Ed. 2010.
2. Selected Topic in Inorganic Chemistry, Mallick, Madan and Tuli, S. Chand Publisher. 17th Ed. 2010.
3. Mehrotra R.C. And Singh, A. Organometallic Chemistry, New Age International Publishers, 2nd Edn, 2000.
4. Gupta B. D. And Elias A. J., Basic Organometallic Chemistry, 2nd Edn., University Press (2013).




