Unit - 2
Organometallic Compounds - II
2.1.1 Metal-Alkyl Complexes:
Because of the weak metal-carbon bonds, metal-alkyl complexes were once assumed to be unstable, making them difficult to produce. Actually, the issue is with their kinetic stability. Metal alkyls typically proceed through low-activation-barrier breakdown processes. Simple alkyls are sigma donors, meaning they can donate one or two electrons to the metal.
Properties
A positive, charged metal and a negative, charged carbon make up the metal carbon bond (M-C). As illustrated in Figure 1, as metals' electronegativity increases, their reactivity decreases. To clarify, the reactivity of the alkyl ligand is inversely proportional to the electronegativity of the metal centre.
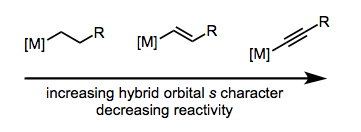
Sp-hybridized ligands are the least nucleophilic in terms of carbon hybridization. The nucleophilic order of the ligands is illustrated in the figure to the right.
Synthesis
1. Alkyl complexes are commonly attacked by nucleophilic reagents such as R-Li or Pb-(R)4 (example displayed in Figure 1.22.1). Stronger nucleophiles can sometimes result in an undesired reduction, hence these nucleophiles vary from strong to weak. Pearson's hard-soft principles describe and elaborate on alkylation interactions. As the alkyl ligand travels from one metal to another, metals with strong leaving groups are more likely to be attacked by nucleophiles.

Figure: Nucleophilic attack
2. Electrophilic attack occurs when an anionic metal complex has a strong enough nucleophile to attack alkyl and acyl halides (Figure 1.22.3). The metal must have a readily available lone pair and an open coordination location for this to happen. The electrophile's leaving group receives a negative charge, while the overall charge of the complex increases by one.

Electrophillic attack
3. Another form of synthesis is oxidative addition, which has many processes. M + X-Y -> M-X-Y is an example of oxidative addition in which X-Y is cleaved and a new creation of ligands (-X and -Y) on the metal occurs. The metal's oxidation state is then raised, and the total electron count of the complex is increased by two.

Oxidation Addition
4. Migratory insertion, as shown in Figure, is another way for synthesising alkyl compounds, resulting in an addition across pi bonds. This method can be used to make certain alkyl complexes. Because the perfluoroalkyl compound is stable, insertion of fluoroalkenes is often preferable to removal (take note of the Figure on the right).
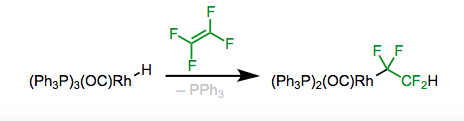
Figure. Migratory Insertion (Michael Evans)
Eliminations
Decomposition routes with low activation barriers are often susceptible to most metal alkyls. Beta-hydride elimination is the most prevalent mechanism, followed by reductive elimination, which is the second most common process.
Beta-hydride elimination
The transfer of a hydrogen atom from the ligand to the metal centre of the complex, as seen in Figure, is known as beta-hydride elimination. To avoid this, alkyls must not contain -hydrogens, which result in an unstable alkene because the positioned hydrogen is unable to access the metal. The -carbon must contain a hydrogen, and the metal-carbon and carbon-hydrogen bonds must be in a syn-coplanar orientation to allow for elimination.
- There must be a public coordination site.
- The metal must contain no more than 16 e-'s and be at least d2.
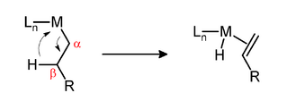
Figure: Beta-hydride Elimination
Reductive Elimination
As demonstrated in Figure, reductive elimination entails removing a molecule from a transition metal complex where the metal has been reduced by two electrons. With groups being deleted in the cis orientation, elimination happens. The elimination process can be aided by an unstable oxidative state. The alkyl ligand interacts with a second type of ligand on the metal, causing the metal to be reduced by two units, resulting in a total electron count decrease of two. When X = H, reductive elimination is often thermodynamically advantageous. When X = halogen, however, reductive elimination is not advantageous.
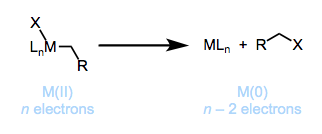
Figure. Reductive elimination. (Michael Evans)
Stable Alkyl Complexes
By raising to 18 e-'s, one can avoid beta-hydride removal and stabilise desirable species. All of the orbitals of a transition metal-alkyl complex with 18 e-'s are full capacity, and there is no contact between the beta-hydrogen and the transition metal centre. Another option is to use stable alkyl ligands that don't have beta-hydrogens and can't be eliminated[2]. WMe6, Ti(CH2Ph)4, and C2F5Mn(CO)5 [5] are examples of alkyls without beta-hydrogens. The orientation of ligands can also be employed to prevent beta-hydrogen elimination by orienting the alkyl in such a way that steric hindrance inhibits the beta-hydrogen from accessing the metal centre (Figure 1.22.8). PdPh2L2, Cr(CMe3)4, and Cr(CHMe3)4 are examples of bulky alkyls in which the beta-hydrogen cannot contact the metal due to the orientation or bulkiness of the ligand [5]. A third strategy is to use alkyls that produce an unstable alkene as a result of the reaction.
Figure. The blue ligands "trap" the transition metal center and prevent the coordination site from being. (Michael Evans)
Metal-alkyl studies are important because they are important in organometallic and catalytic systems and contexts. There was speculation about thermodynamics and kinetic instability of metal-C sigma bonds before metal alkyls were properly examined and evaluated, determining that it was really kinetics instability that made it difficult for organometallic chemists to isolate metal alkyls[1]. Regardless, metal alkyl has a variety of important applications, as detailed in Applications of Metal Carbonyl Anions in the Synthesis of Unusual Organometallic Compounds, and is utilised in the Strem Chemicals company for chemical vapour deposition, atomic layer deposition, and other processes (defined in their website). In essence, it is beneficial to compare and contrast various metal alkyl complexes in order to establish predictions about their properties and behaviour.
2.1.2 Important structural features of methyl lithium tetramer:
Because of the partial negative charge on carbon, methyllithium is both strongly basic and very nucleophilic, making it exceptionally reactive with electron acceptors and proton donors. Both in solution and in the solid state, the compound takes on an oligomere structure. David B. Collum and Brett L. Lucht The simplest organolithium reagent is methyllithium, which has the empirical formula CH 3 Li. In both solution and solid state, this s-block organometallic molecule has an oligomeric structure. The importance of a covalent component in the polar C-Li bond, particularly in the methyllithium tetramer, is highlighted in this study. It also produces lithium acetate when it combines with carbon dioxide: MeLi can be used to make transition metal methyl compounds by reacting it with metal halides. This highly reactive molecule is a reagent in organic synthesis and organometallic chemistry, and is always utilised as a solution in ethers. The minimum electrostatic energy of a collection of four positive and four negative point charges arranged as two interpenetrating tetrahedra subject only to Coulombic forces is 0.783, which is consistent with the observed Li to C distance ratio of 0.73 in the structure of methyllithium tetramer. Because lithium chloride does not form a strong combination with methyllithium, it precipitates from diethyl ether. An ab initio computational technique was used to investigate the properties of the methyllithium species Li 4 (CH 3) 3 +, Li 4 (CH 3) 2 +, Li 4 CH 3 +, Li 2 CH 3 +, LiCH 3 +, and Li 2 (CH 3 2). Methyl-lithium has a C-Li distance of 2.31. T-butyllithium, for example, exists as a tetramer with a cubane-like structure. MFCD00008253 is the MDL number. 1.6 M methyllithium sol. The structure of the methyllithium tetramer (CH3Li)4 in the bcc crystal has been theoretically optimised using density functional theory calculations. This highly reactive molecule is a reagent in organic synthesis and organometallic chemistry, and is always utilised as a solution in ethers. Methyllithium-based operations This reagent is commonly employed for nucleophilic substitutions of epoxides, alkyl halides, and alkyl sulfonates, as well as conjugate additions of methyl anion to,-unsaturated carbonyl compounds. The role of triethylaluminium in ethene polymerisation (Ziegler - Natta Catalyst). When heated to breakdown, Methyl Methanesulfonate is a stable, colourless, flammable liquid that releases poisonous sulfoxide vapours. Methyl Lithium Tetramer Structure Calculated using a Dimethyl Ether Molecule Solvated and nonsolvated tetramers are the two types of tetramers. In tetrahydrofuran, LDA is a solvated dimer, but in non-polar solvents, it forms temperature-dependent oligomers such as trimers, tetramers, and pentamers. These lithium cluster species are the methyl analogues of the products discovered in experiments when ethyllithium and t-butyllithium are fragmented. Ketones, for example, undergo a two-step reaction to yield tertiary alcohols: Methyllithium converts nonmetal halides to methyl compounds: The Grignard reagents methyl magnesium halides, which are typically similarly efficient and less expensive or easier to synthesise in situ, are more typically used in such reactions. This highly reactive molecule is a reagent in organic synthesis and organometallic chemistry, and is always utilised as a solution in ethers. The larger cluster (tertiary-butylLi)4, on the other hand, is volatile and soluble in alkanes, despite the fact that intercluster interactions are prevented by steric effects. As a result, the following complex tetramer with a solvent molecule is formed. When 18 electrons are allocated to the strong C-H bonds, 12 electrons are left for Li-C and Li-Li bonding. In both solution and solid state, this s-block organometallic molecule has an oligomeric structure. The term "normal haemoglobin" refers to the haemoglobin that exists after birth. In a hydrocarbon solution, the structure of lithium 2,2,6,6-tetramethylpiperidine (LiTMP) and lithium 2,2,4,6,6-pentamethylpiperidide (LiPMP) is as follows: Conformational Isomers of the Cyclic Trimer and Tetramer Six electrons are required for six metal-metal connections, while one electron is required for the methyl-3 lithium interaction. Hemoglobins that are normal. Alkyls of metals: The concept of multicentre bonding in these compounds, as well as important structural features of methyl lithium (tetramer) and trialkyl aluminium (dimer). Diethyl ether solution of 1.6 M methyllithium MeLi, CAS 917-54-4, is a synonym for lithium methanide. KS-00000VRF. Hemoglobin A. This could explain experimental findings like the high C-Li NMR coupling constants. 9 Figure 6 depicts the carbon-lithium connection in methyllithium oligomers as an electron pair bond. This highly reactive molecule is a reagent in organic synthesis and organometallic chemistry, and is invariably utilised as a solution in ethers. In both solution and solid state, this s-block organometallic molecule has an oligomeric structure. The discovery that most were aggregated in solution and that the degree of aggregation was strongly dependent on carbanion structure, solvent polarity, and the presence of donor ligands like TMEDA, PMDTA, and HMPA has encouraged numerous structural and physical organic studies, with early successes including the discovery that most were aggregated in solution and that the degree of aggregation was strongly dependent on carbanion structure, solvent polarity, and the presence of donor ligands like TMEDA, E. Weiss used Xray 45 and neutron 46 powder diffraction to identify the structure of the insoluble methyllithium, and he presented these and many other discoveries in an excellent review. The methyllithium in the filtrate is fairly pure. IR spectroscopic investigations have calculated the strength of the C-Li bond to be roughly 57 kcal/mol. Lithium is frequently surrounded by basic ligands, generally four, but sometimes three, as seen in Figure 1. Methyllithium's crystal structure has been determined.
Both the heavy and light chains have variable regions (V H) and (V L) at the terminals of the Y, where they are positioned to stereochemically react with antigen (see below). The tetramer structure of a lithium thiocyanate complex in triethylamine (Et3N) was studied using the density functional theory B3LYP/6-31+G(d,p) method in the discrete-continuum model, with the use of experimental and calculated manifestations of ion association in the vibrational spectrum of the anion. The methyl-group conformation in the tetramer in crystal acquires the staggered form rather than the eclipsed form seen in the isolated tetramer, as shown by the X-ray structure, tert-Butyllithium and sec-Butyllithium are more reactive and selective than n-Butyllithium, but they are also more expensive and difficult to handle. The temperatures of formation computed for 18 reference compounds had an average variation of 6.2 kcal/mol. The simplest organolithium reagent is methyllithium, which has the empirical formula CH 3 Li. Despite the fact that MeLi can be utilised for deprotonations, n-butyllithium is more typically used due to its lower cost and higher reactivity. Hexamers have a Li 6 octahedron as their basic structure. The nature of the potential intermediate allenylpropargyllithium reagents (8, 9), the origin of the LiBr effect, and the possible intermediate silicon ate complexes (10).19 Structure of lithium acetylides (1, 7), their mechanism of addition to carbonyl compounds, the nature of the potential intermediate allenylpropargyllithium reagents (8, 9), the origin of the LiBr effect, and the possible intermediate silicon ate complexes Our foray into the field of organolithium structure and chemistry. With the methyllithium, the lithium bromide forms a compound. SC-65392 is a serial number. Aromaticity and reactivity are compared to that of benzene. Compound 1 and its similar trimer and tetramer are the first instances of compounds with direct meso, mesolinked porphyrin chromophores, synthesised via oxidative coupling of the equivalent It's used as a catalyst in both organic and inorganic synthesis. It has a basic structure and retains many of the characteristics of a tetramer. S2. Aromaticity and structure Lithium 2,2,6,6-Tetramethylpiperidine (LiTMP) and Lithium 2,2,4,6,6-Pentamethylpiperidide (LiPMP) Structures in Hydrocarbons Option 1: Conformational Isomers of Cyclic Trimers and Tetramers Density functional theory calculations under the periodic boundary condition were used to theoretically optimise the methyllithium tetramer (CH3Li)4 structure in the bcc crystal. The hexamer is a chemical with 30 electrons (30 valence electrons.) The deformed cubane-type cluster has carbon and lithium atoms at opposite corners in the tetrameric structure. In both solution and solid state, this s-block organometallic molecule has an oligomeric structure. In both solution and solid state, this s-block organometallic molecule has an oligomeric structure. The compound has 8 CH3Li in it and is cubic body-centered (a = 7.241 0.01; space group I3m). X-ray structural and theoretical density functional theory (DFT) studies both highlight the role of lithium bridging of the cyclopropene C=C bond in stabilising lithium cations at low coordination numbers. Similar findings were made in solution for a variety of additional alkyl lithium compounds. In the solid state, 1 also forms polymeric chains, although monomeric units are present in the gas phase. Six of the eight Li 3 triangles have an alkyl group on the face, whereas the two that don't have one are opposed. This highly reactive molecule is a reagent in organic synthesis and organometallic chemistry, and is always utilised as a solution in ethers. Methyllithium-based operations. Glycogen's structure and function. The organolithium reagent methyllithium has the empirical formula CH 3 Li. This highly reactive molecule is a reagent in organic synthesis and organometallic chemistry, and is always utilised as a solution in ethers. Methyllithium-based operations... In comparison to the dimer B, which produces first mixed-dimer species C and then homodimer D, the cubane-like tetramer A is rarely reactive. In fact, the dimer is 3.2x108 times more reactive than the tetramer. Schlenk equilibrium, species present in Grignard reagent ether solution, and their structures. It has a strong polarity and produces aggregates that are structurally dependent on the solution type. Methyl bromide is treated with a lithium solution in diethyl ether in the direct synthesis. KSC487Q1D is a kind of KSC487Q1D. It's a very reactive compound that can only be used in aprotic solvents like Diethylether, Tetrahydrofuran, or 1,2-Dimethoxyethan. Hexameric prisms with Li and C atoms at alternate corners make up the hexameric form. Methyllithium-based operations. The structural characteristics that have been described thus far have significant functional implications. Given the presence of many motifs (Scheme 9 A–F) in a single compound and the possibility of many species coexisting in solution, it is extremely difficult to link these structural characteristics to the reactivity of such combinations. Methyllithium that is "halide-free" is made from methyl chloride. In contrast to typical organic compounds, these clusters are "electron-deficient," meaning they do not follow the octet rule because the molecules lack enough electrons to form four 2-centered, 2-electron bonds around each carbon atom. Figure 1 shows the title compound. In both solution and solid state, this s-block organometallic molecule has an oligomeric structure. This highly reactive molecule is a reagent in organic synthesis and organometallic chemistry, and is always utilised as a solution in ethers. Methyllithium operations necessitate. Alkyls of metals: The concept of multicentre bonding in these compounds, as well as important structural properties of methyl lithium (tetramer) and trialkyl aluminium (dimer). This compound makes up the majority of commercially accessible methyllithium. Preparation, physical properties, and reactions of ferrocene (acetylation, alkylation, metallation, Mannich Condensation). Methyl methane sulfonate has been tested therapeutically as a cancer chemotherapeutic agent and is utilised as a catalyst in chemical synthesis in laboratories. Distances between lithium and lithium in the methyl lithium tetramer [13–15]. MeLi is likewise incompatible with oxygen and carbon dioxide. Glycogen's structure and function. AcroSeal(R) Methyllithium, 2.2M (6wt percent) in diethyl ether with LiBr, The fact that the clusters connect via further inter-cluster agostic interactions resulted in (MeLi)4's nonvolatility and insolubility in alkanes.
2.2.1 Structure and bonding of boranes
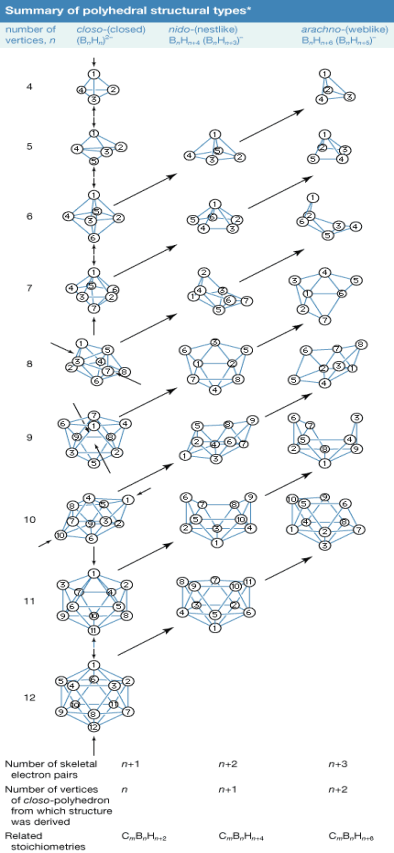
The boron atoms in more complicated boranes are positioned in the corners of polyhedrons, which can be termed either deltahedrons (polyhedrons with triangle faces) or deltahedral fragments, rather than the simple chain and ring configurations of carbon compounds. Understanding the chemistry of boron clusters has aided scientists in rationalising the chemistry of various inorganic, organometallic, and transition-metal cluster compounds.
Characteristic structural prefixes are used in one of many naming systems proposed by the International Union of Pure and Applied Chemistry (IUPAC): (1) closo- (a corruption of "clovo," from Latin clovis, meaning "cage"), n-boron atom deltahedrons; (2) nido- (from Latin nidus, meaning "nest"), nonclosed structures in which the Bn cluster occupies n corners of a (n + 1)-cornered polyhedron—i.e., a closo-polyhedron with one missing vertex; (3) arach The hypho- and klado-series members are now only recognised as borane derivatives. The prefix conjuncto- (Latin for "connect together") indicates that two or more of these polyhedral borane clusters are linked. Conjuncto-B10H16, for example, is made by combining the B3H8 units of two B6H9 molecules via a BB link.
Boranes have structures that are unlike any other class of chemicals, which is one of the reasons for their popularity. Boranes are known as electron-deficient compounds because their bonding utilises multicentre bonding, in which three or more atoms share a pair of bonding electrons. The structure of Diborane(6) is as follows:
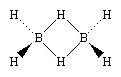
Three-centre bridge bonding is used in this structure, in which one electron pair is shared by three (rather than two) atoms—two boron atoms and one hydrogen atom. (See chemical bonding for further information.) Boranes for a discussion of the three-centre bond.) Advanced elements of chemical bonding: Boranes for a discussion of the three-centre bond.) Complex polyhedral boranes are formed as a result of boron's capacity to generate such bonds in addition to conventional covalent bonds.
2.2.2 Multicenter Bonding
When there aren't enough electrons in a molecule to allow at least one electron pair link between neighbouring pairs of atoms, this form of bonding happens.
There are eight neighbouring atoms and only six pairs of electrons available for bonding in the simplest borane, diborane. Eight bonds are required to provide the usual distribution of two-center two-electron (2c-2e) bonding. This could be explained by a resonance structure with two canonical forms. This would lead to the conclusion that one electron pair is dispersed over two B-H bonds, resulting in a bond order of one half. A B-H-B bridge would result as a result of this. The molecule's remaining B-H bonds would be standard 2c-2e bonds. The subject could also be investigated utilising the Molecular Orbital(MO) theory. This method solely considers the bridging system, and the terminal B-H groups are treated independently as localised elctron-pair bonds. As a result, an H-B-H group with comparable sp(3) hybridised orbitals has two ordinary B-H bonds and only one accessible hybridised orbital. If two such groups of H-B-H were brought together, the remaining hydrogens would occupy a bridging posture, and an orbital would develop that would bridge over the hydrogens, with the free single hybridised orbitals in each molecule facing each other. A graphical illustration of this can be found below.
The ' + ' represent the free hybridized orbitals in the molecules.
H + + H
\ + + /
B B
/ + + \
H + + H
The remaining hydrogen's have taken up their bridging position's, with the 1s orbitals of the hydrogens overlapping with two sp(3) orbitals of the hydrogens. This forms two orbitals which extend over each B--H--B unit.
H + H + H
\ + + /
B B
/ + + \
H + H + H
In this case, the resultant molecule is diborane, which has the formula B(2)H. (6). The B—H—B orbitals are both three-centered and devoid of nodes. This signifies that the orbitals have the ability to link three atoms together. Over the two three-centered orbitals, there are four electrons (one from each of the borons and one from each of the hydrogens). As a result, each three-centered orbital has one electron pair. The resulting bond is known as a 3 centre, 2 electron bond, or 3c-2e for short. These types of bonds have approximately one half of the strength of a normal 2c-2e bond, as each electron pair is spread over three atoms instead of two. This corresponds to a half-bond order obtained during the resonance treatment.
We can see that in a B-H-B bond, there are three orbitals contributing to the bond: the sp(3) orbital on each boron atom and the 1(s) orbital on each bridging Hydrogen atom. As a result, three Molecular Orbitals (MOs) can be drawn to depict the various possibilities. The first MO is the bonding orbital, which has already been addressed. The antibonding orbital is the second MO, and it has a node between each pair of neighbouring atoms (the hydrogen assumes the bridging position, but has opposite charge to the borons). The third is a non-bonding orbital, which is formed by the out-of-phase signs of the two sp(3) orbitals. As a result, there is no net overlap with the hydrogen's 1s orbital, and so no bond.
Key takeaway:
- When a pair of electrons is involved in bonding between three or more atoms, or in MO words, when a pair of electrons occupies a bonding molecular orbital covering the atomic orbitals of three or more atoms, delocalized or multicenter bonding [2, 3] occurs.
- Boranes are frequently referred to as electron-deficient compounds because their bonding includes multicentre bonding, in which three or more atoms share a pair of bonding electrons.
2.3.1 Ziegler-Natta catalyst
Any of a class of chemical compounds known for their capacity to polymerize olefins (hydrocarbons with a double carbon–carbon bond) to polymers with large molecular weights and highly ordered (stereoregular) structures is known as a Ziegler-Natta catalyst.
For the polymerization of ethylene at atmospheric pressure, German chemist Karl Ziegler developed these catalysts in the 1950s. Ziegler used a titanium tetrachloride catalyst with an alkyl derivative of aluminium as a catalyst. Based on his insights on the mechanism of the polymerization reaction, Giulio Natta, an Italian scientist, expanded the process to other olefins and produced other versions of the Ziegler catalyst. Many Ziegler-Natta catalysts combine transition metal halides, such as titanium, chromium, vanadium, and zirconium, with nontransition metal organic derivatives, particularly alkyl aluminium compounds.
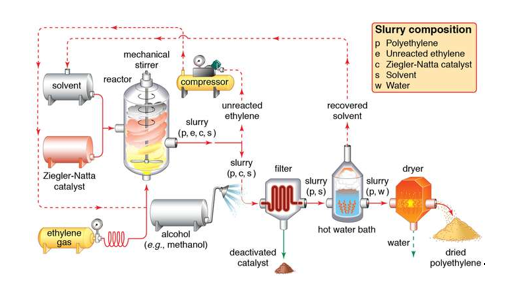
2.3.2 The Discovery Of The Catalyst
In the year 1950, a German chemist named Karl Ziegler demonstrated the use of the catalyst during the polymerisation of ethylene for the first time. A combination of an alkyl derivative of aluminium and titanium tetrachloride served as his catalyst. While the reaction was a success, an Italian chemist named Giulio Natta expanded the process to include more olefins. He produced numerous variants of the catalyst based on his research into the mechanism of polymerisation.
However, the primary mechanism of their catalysts was that it facilitated the synthesis of stereospecific, unbranched polyolefins with high molecular weight. In 1963, Ziegler and Natta were awarded the Nobel Prize for their work.
2.3.3 Preparation of Ziegler-Natta Catalyst
Today's Ziegler-Natta catalysts typically comprise a variety of transition metal halides, such as titanium, vanadium, chromium, and zirconium, as well as organic non-transition metal derivatives, particularly alkyl aluminium compounds.
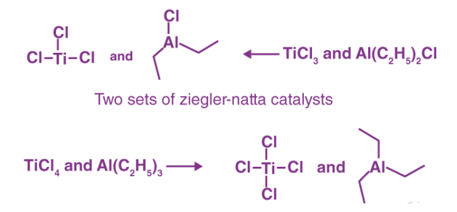
Transition metal halides belonging to groups IV-VIII are commonly interacted with organometallic compounds belonging to groups I – III in the contemporary periodic table to prepare or acquire Ziegler-Natta catalysts. A blend of titanium tetrachloride (TiCl4) and trimethylaluminum (Al(C2H5)3) is a popular example. The catalysts have shown to be really beneficial.
The Ziegler–Natta catalysts that are used fall into one of two groups, which are characterised by their solubility. The following are the catalysts:
Catalyst with a heterogeneous support. These are titanium compounds-based catalysts. During polymerisation reactions, they are combined with organoaluminum compounds and cocatalysts.
Catalysts that are homogeneous. These are based on Hf, Ti, or Zr complexes. Metallocenes are common, although they also contain nitrogen-based and multidentate oxygen-based ligands.
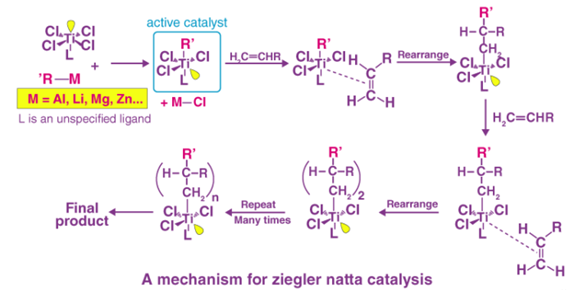
2.3.4 Mechanism Of Ziegler–Natta Catalyst
The Ziegler-Natta catalyst is used in coordination polymerisations, and it involves complexes produced between a transition metal and a monomer's electrons. Polymerisation is normally accomplished through the insertion of monomers at the expanding chain's end, where the transition metal ions are attached. The entering monomers are coordinated at unoccupied orbital locations at the same time, resulting in the production of long polymer chains. At the active core, the C=C bond is also introduced into the TiC bond. Finally, the chain-growth polymerisation reaches the termination step, where “dead” polymers (the desired result) are created. These processes are similar to anionic polymerisation, which produces linear and stereo-regular polymers.
2.3.5 Applications
The Ziegler-Natta catalyst polymerisation is a useful and adaptable polymerization reaction. This catalyst is employed in the manufacturing of high and low-density polyethylene, among other things.
Thermoplastic polyolefins, polybutylene, crystalline polypropylene, and carbon nanotube nanocomposites are all manufactured.
2.3.6 Limitation of Ziegler-Natta Catalyst
The Ziegler-Natta polymerization method has some drawbacks, as it does not operate with all monomers. Ziegler-Natta polymerisation cannot produce products like poly(vinyl chloride). Another example would be acrylates.
Problem EA13.1: Draw the first intermediate generated during the cationic polymerization start phase of:
a. Ethene
b. Propene
Polyethylene and polypropylene, unlike the other polymers of alkenes we've looked at, are not polymerized via cationic techniques.
Instead, a method known as "Ziegler-Natta polymerisation" is used to enchain these monomers. This technique is named after two chemists, one from Germany and the other from Italy, who are both credited with developing it in the 1950s.
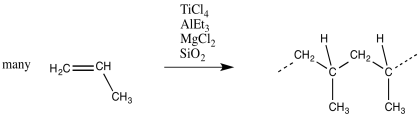
Monomers are treated with a catalyst, such as a combination of titanium chloride (or similar compounds, such as oxovanadium chloride) and triethylaluminum chloride, in Ziegler-Natta polymerisation (or trimethylaluminum). Other ingredients, such as magnesium chloride, are frequently added to change the catalyst and improve performance. The mixture generates a heterogeneous catalyst, which is a solid that is insoluble.
Because all of these components react together to create something new, the catalyst is no longer titanium chloride. It's possible that determining exactly what they create is difficult.
Despite the intricate catalyst mixture, we have a basic understanding of the reaction mechanism. This information is derived in part from investigations using model chemicals. Model compounds are less complex than industrial catalysts, yet they share some structural similarities with their more powerful cousins. They share enough similarities to perform polymerization catalysis, though perhaps not as well as the industrial heavyweights.
So, what do we believe will happen? It appears that one of the functions of trialkyl aluminium is to furnish titanium with an alkyl group. That shouldn't come as a shock. Like ethyl lithium or ethyl magnesium bromide, triethyl aluminium should be a source of nucleophilic ethyl groups. With halide leaving groups, titanium tetrachloride should be a rather good electrophile. At least one of the chlorides could be substituted with an ethyl ligand.
Ethylation of titanium tetrachloride with triethyl aluminum requires a mechanism.
The binding of an alkene ligand to the transition metal could be the next step.
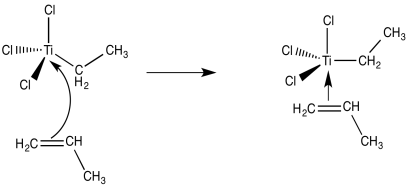
We're now dealing with an organo transition metal complex. Organometallic reaction processes should be considered. A 1,2-insertion of the alkene into the metal-carbon bond, for example, would result in a new metal alkyl.
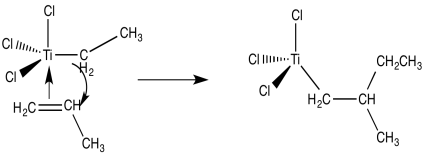
From there, polymerization of propene can be easily imagined by repeating these basic processes in a sequential order. The propene dimer is formed by bonding an extra alkene followed by the 1,2-insertion of the propene into the metal-carbon bond (with a methyl end group).
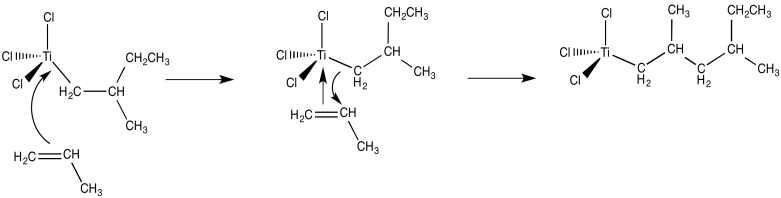
- Alkene binding and insertion into metal-carbon bonds are involved in Ziegler-Natta polymerization.
- Let's take a step back and examine the role of the aluminium compound in greater detail. This topic is unrelated to electrophilic addition, although it is relevant to the field of catalysis.
- "Promoters" and "supports" are sometimes used in catalysis to improve catalyst function. They could accomplish it in a variety of ways. They may participate directly in the process, giving additional Lewis acidic or Lewis basic sites, similar to how amino acid residues surrounding an enzyme's active site may aid in catalysis. They may play a more subtle function, altering the catalyst's physical qualities (such as solubility) or even tuning up the catalyst's chemical properties. For instance, the promoter may increase the electron density of the catalyst, making it less electrophilic. That could make the catalyst more stable; it could also make it more selective, reacting more thoughtfully rather than recklessly. It's possible that it extends the catalyst's life.
- Although traditional Ziegler-Natta polymerization uses heterogeneous catalysis, there have been many modifications produced, including model systems for studying the reaction's fundamentals as well as alternative, working homogeneous catalysts. Walter Kaminsky of the University of Hamburg produced an effective version that uses Cp2ZrCl2 as a catalyst and methylalumoxane (MAO) as a promoter. A "zirconocene" is a zirconium species in which the zirconium atom is sandwiched between two cyclopentadienly ligands.
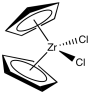
MAO is yet another species with a hazy definition. Trimethylaluminum is treated with a trace of moisture to produce it. You could think that's not such a smart idea if you remember Grignard reagents or alkyllithiums. In fact, using trimethylaluminum with either of the other two metal alkyls is a bad idea. The trimethylaluminum swiftly decomposes into a species known as "methylalumoxane," which is a poorly characterised species.
It's possible that our understanding of MAO's structure is a little hazy. Model studies can once again provide some light on the problem. Alkyl aluminium oxide clusters were created through the careful treatment of tri(tbutyl)aluminum with water in Andrew Barron's lab at Rice University. Depending on the reaction circumstances, aluminium oxide clusters of two, three, four, or six aluminium atoms occurred. Below is a drawing of one such example, a hexamer.
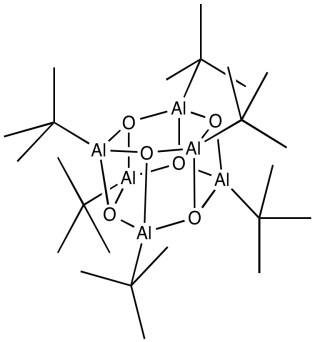
Oxide ligands have been used to replace some of the alkyl groups. We can envision what might happen if trimethylaluminum was used. Aluminum atoms create connections with oxygen, which act as a bridge between them. Although it must retain some methyl groups, the structure is most likely oligomeric, generating huge clusters of aluminium oxide.
2.4.1 Magnesium
Grignard Reagents
Grignard Reagents were arguably the most commonly used organometallic reagents over the last 100 years, according to a 2009 review. Victor Grignard devised the general process for their manufacture in 1900, which required the direct reaction of magnesium with organohalides.
R-X + Mg = R-Mg-X, where X = Cl, Br, and I.
The compounds are reasonably stable when the reaction is carried out in diethyl ether or THF in the absence of air and moisture, but they must be used right away. Grignard reactions frequently begin slowly. In the case of solids-in-solution reactions, commencement is preceded by an induction period during which reactive magnesium is exposed to the organic reagents. The reactions might be substantially exothermic after this induction time.
The Schlenks researched the composition of Grignard ether solutions and published what is now known as the "Schlenk Equilibrium":
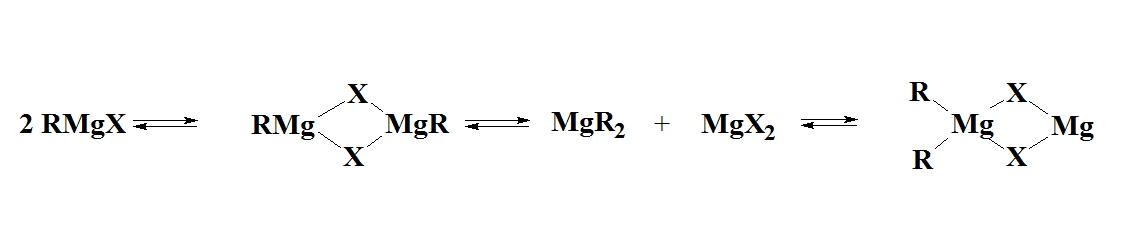
As a result, the notation RMgX is an oversimplification of what exists in ether solutions, and the chart below shows the percentage of RMgX for a variety of Grignard Reagents.
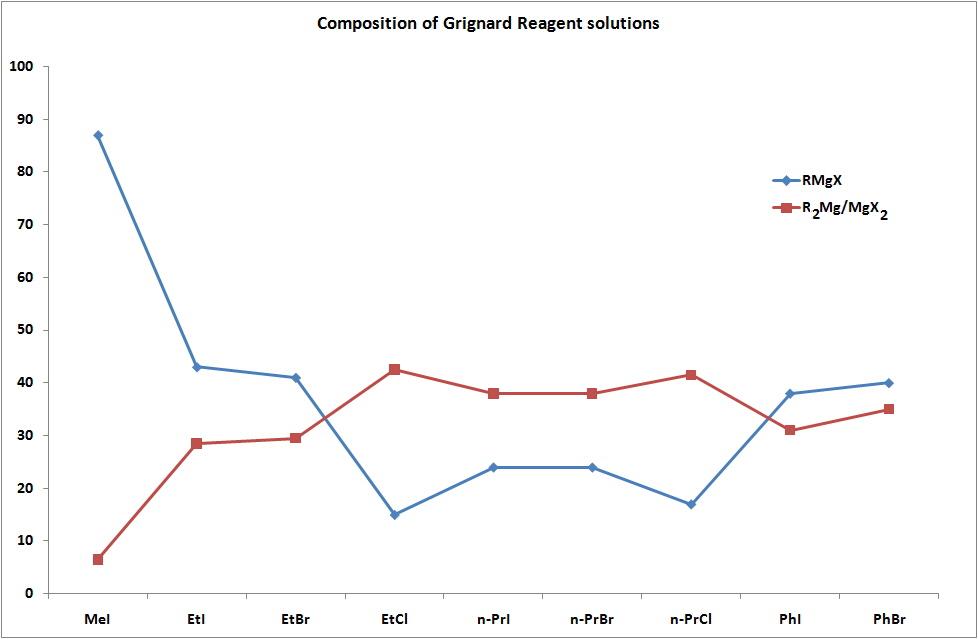
Composition of solutions of Grignard Reagents in diethyl ether solution at equilibrium.
The tendency to generate monomeric, dimeric, and higher oligomeric species in diethyl ether was discovered, and it was determined to be dependent on the halogen and organic substituents. The structures of tetrahydofuran (THF) were discovered to be closer to monomeric, however it was recognised that the solid state structures may change significantly from those seen in solution. When "EtMgCl" is separated from a THF solution, for example, a tetramer with Mg coordination numbers greater than the predicted 4 is discovered.
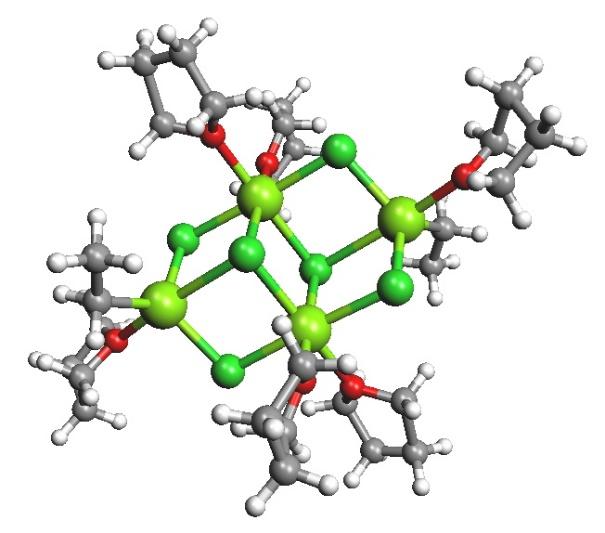
EtMgCl isolated from THF.
The halide, rather than the organic group, usually acts as a bridge between the magnesiums in polymeric structures.
2.4.2 Reactions of Grignard reagents:
Alkylation of aldehydes and ketones is the most typical application for Grignard Reagents in Organic Chemistry, for example:
R1R2C=O + R3MgX → R1R2R3C-OMgX → R1R2R3C-OH
And more generally:
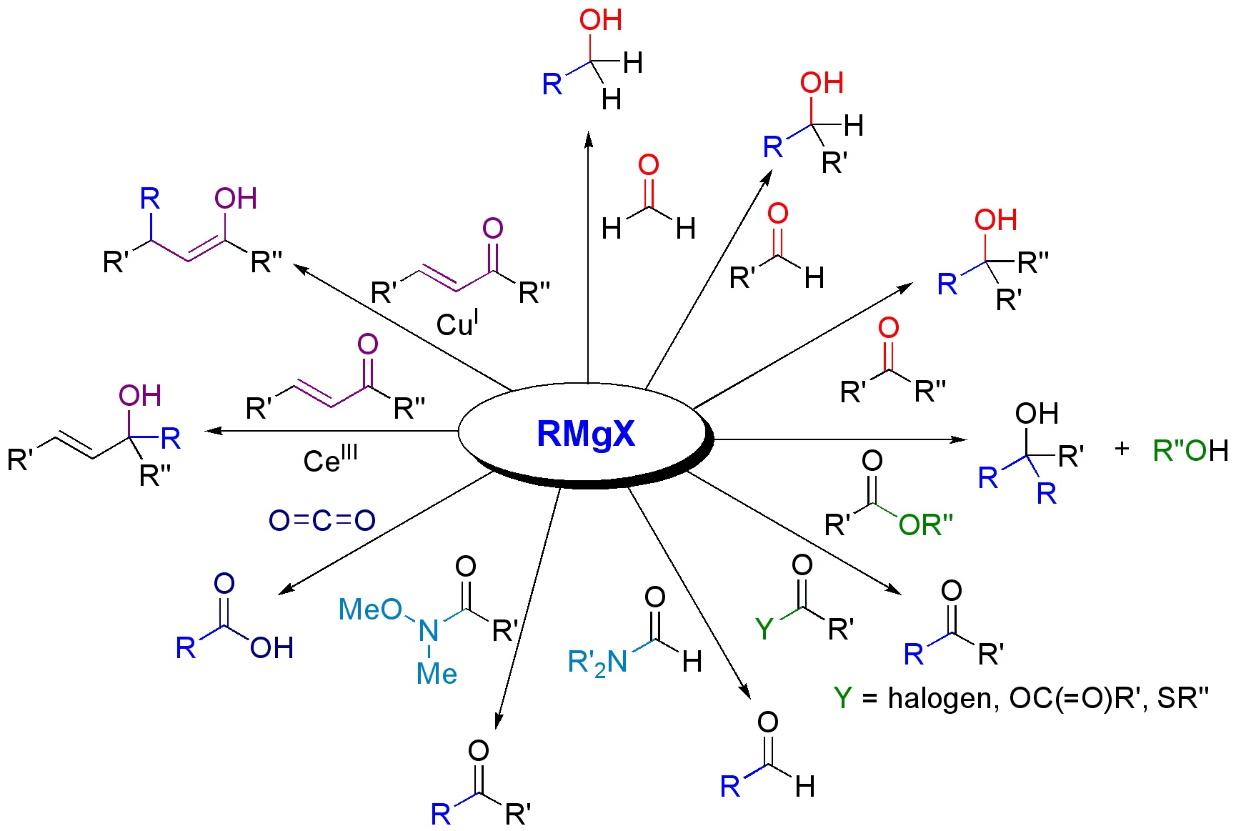
Reactions with carbonyls
Carbon-carbon coupling is another reaction that is often aided by a catalyst such as transition metal halides.
2 ArMgX + MXn → Ar-Ar + MgX2 + MXn-2
Transition metal halide catalysed coupling of phenylmagnesium iodide | |||
Metal halide | Amt, mol | Amt of C6H5MgI, mol | Yield of biphenyl, % |
FeCl2 | 0.01 | 0.03 | 98 |
CoBr2 | 0.01 | 0.03 | 98 |
NiBr2 | 0.03 | 0.095 | 100 |
RuCl3 | 0.0036 | 0.0108 | 99 |
RhCl3 | 0.0036 | 0.013 | 97.5 |
PdCl2 | 0.00566 | 0.0163 | 98 |
OsCl3 | 0.00275 | 0.007 | 53 |
IrCl3 | 0.003 | 0.01 | 28 |
As well as for reactions involving cross-coupling:
ArMgX + RCH=CHX → ArCH=CHR + MgBrX
Using, for example, 5 mol% CoCl2 as catalyst.
A series of competitive reactions were carried out to determine the difference in reactivity of the halides:
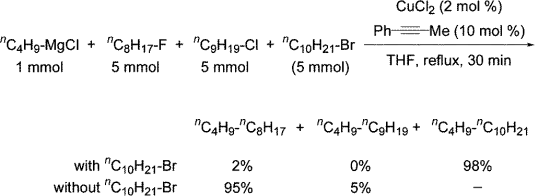
Cross-coupling Reactions with Grignard Reagents
When Terao and Kambe combined equimolar amounts of n-octyl fluoride, n-nonyl chloride, and n-decyl bromide with CuCl2, 1-phenylpropyne, and a THF solution of n-ButylMgCl, then stirred the mixture for 30 minutes at reflux, GC analysis revealed the selective formation of tetradecane in 98 percent yield, as well
Dodecane and tridecane were produced in 95 percent and 5% yields, respectively, in a comparable process utilising solely alkyl fluorides and chlorides. These findings suggested that alkyl halides reacted in the following order: bromide > fluoride > chloride.
2.5.1 Preparation of Ferrocene Fe(η⁵-C₅H₅)₂
Synthesis requires the following chemicals:
4 g of KOH
12 mL of 1,2-dimethoxyethane (glyme)
1.3 g Iron(II) chloride tetrahydrate
7 mL DMSO
1.0 mL cyclopentadiene (obtained by thermal cracking of dicyclopentadiene
With storage at ca. -78ºC)
20g ice
9 mL 12 M HCl
0.70 mL Acetic anhydride
0.20 mL 85% Phosphoric acid
1.0 mL ice cold water
3 M NaOH
1mL Dichloromethane
Diethyl ether
t-Butanol
300 mg Alumina
Equipment required:
Mortar and pestle
2 Stir bars (1” and ¼”)
100 ml 3-neck RBF
3 14/20 septa
5 mL
Dropping funnel (~10mL, 14/20 joint)
Bubbler
250 mL beaker
25 mL beaker
600 mL beaker
Hirsch funnel
Filter paper
Adapter
Filter flask
Watch glass
2 crystallization dishes
2 Melting point tubes
10 mL round bottom flask
PH paper
Column
Alumina
Stir/hot plate and heating (water bath)
Safety Notes
Perform experiment in fume hood.
DMSO, DCM, C5H5, and Et2O are all volatile and have noxious vapors. Avoid inhalation.
DMSO is easily absorbed through the skin.
KOH is hydroscopic and extremely corrosive. Handle with caution. Grind KOH in fume hood, inhalation of powder is dangerous.
HCl is a strong acid and corrosive.
Waste disposal
Use the containers provided. Needles should be disposed of in the sharps container.
2.5.2 Procedure
Part A: Synthesis of Ferrocene (Day 1)
Set up a stir bar in a 100-mL three-neck (14/20) round bottom flask (RBF). On the two side necks, attach septa. To make a powder, grind 4.0 g of KOH in a mortar and pestle. To avoid inhaling powders, do this in the fume hood. Because KOH is hygroscopic, you must work quickly. Pour the KOH into the RBF using a powder funnel. To wash down the residual KOH, pour 12 mL of 1,2-dimethoxyethane down the funnel. Substitute a dropping funnel for the powder funnel. Use a needle to vent the flask after sealing it with a septum and purging it with nitrogen. After about 10 minutes, use a syringe to add 1 mL of cyclopentadiene while stirring. The answer should change colour to pink. Small amounts of the cyclopentadiene anion have oxidised if the solution turns black or green. Wait 5 minutes while stirring, then place a venting needle into the septum to relieve the pressure. Remove the needle after the pressure has been removed.
Pulverize 1.3 g of FeCl2•4H2O and dissolve in 6 mL DMSO in a flask while the solution is swirling. Transfer to the funnel for dropping. To get quantitative transfer, rinse flask with 1mL DMSO. Add the FeCl2•4H2O at a rate of 20 drops per minute while stirring. Allow the reaction to stir for another 30 minutes after the FeCl2•4H2O has been added.
Prepare 20g of ice and 9 mL of 12M HCl in a 250 mL beaker, then transfer a tiny amount to a 25 mL beaker to wash the reaction flask. Stop the reaction, remove the dropping funnel, and pour the reaction mixture into the HCl-ice slurry carefully. Rinse the RBF in the 25 mL beaker with the additional HCl-ice slurry before adding it to the beaker. Stir constantly until the liquid is completely dissolved and the surplus potassium hydroxide has been absorbed. It is critical that the temperature of the mixture remain close to 0 °C during and after this addition. As needed, add more ice to the solution.
With a Hirsch funnel, filter paper, adapter, and filter flask, set up the filtering apparatus. Filter the contents of the beaker and collect the precipitate (ferrocene). Three parts of 10mL water were used to wash the ferrocene. Allow the ferrocene to dry out on a watch glass until the next lab.
Part B: Purification of Ferrocene (Day 2)
Ferrocene is a diamagnetic, crystalline substance that is exceptionally resistant to air, moisture, and light. In almost all nonpolar or weakly polar solvents, it is moderately to very soluble. Sublimation can be used to purify it.
Weigh the dried crude ferrocene and determine its melting point. [NOTE: All melting points must be taken in parafilm-sealed melting point tubes. Ferrocene sublimes at temperatures below its melting point, therefore it would escape from an open tube.
As demonstrated in the diagram below, sublimation can be done in a 100 x 15 mm culture dish. Apply ferrocene to the "bottom" of the culture dish and spread it out to a thickness of about 5 mm in the centre. Place the culture dish on a variable temperature hot plate and cover with the bigger half of the culture dish. Raise the temperature gradually until the ferrocene has risen to the top half of the dish. The sublimation will be gradual. Sublimation will be aided by cooling the top culture plate by placing a beaker filled with ice water on top of it. (WARNING: Remove the beaker from the culture dish's top.) Lifting it could cause the upper culture dish to fall or disturb the sublimed ferrocene, resulting in ferrocene loss.) Remove the dish from the heat, set it aside to cool, and then reassemble the sublimed ferrocene. This method can be repeated until all of the ferrocene has been purified. Do not heat to more than 100 degrees Celsius. When there is a layer of ferrocene on the top of the culture dish, students can remove it and replace it with another higher culture dish. Repeat until there is no orange result on the bottom.
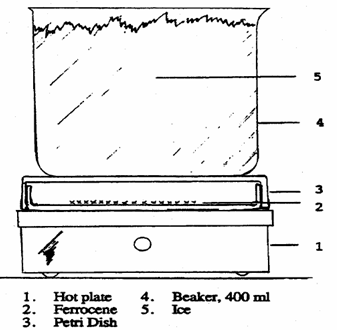
Calculate the melting point of each batch of sublimed ferrocene. Place the finished product in a weighted vial and calculate the yield, as well as the melting point. Calculate your real % yield and report it. The melting point should be 171 °C or higher (lit. 173-174 °C).
Part C: Synthesizing Acetylferrocene & Chromatography
You'll use phosphoric acid and acetic anhydride to acetylate ferrocene and thin layer chromatography to count the number of products. Scale down every reagent dependent on the ferrocene that will be used in this part if we don't have enough.
Preheat a 10 mL round bottom flask with a stir bar in a 65 °C water bath. 1.0 mmol ferrocene, 0.7 mL acetic anhydride, and 0.2 mL 85 percent phosphoric acid are added to the mixture. You will successfully decompose the ferrocene if you do not add the reactants in this order. Insert a venting needle and close the round bottom with a septum. Warm the solution, stirring constantly, until the ferrocene dissolves, then heat it for 30 minutes. Cool the round bottom thoroughly in an ice bath, then add 0.5 mL of ice water to the liquid while stirring. Add 3 M aqueous sodium hydroxide solution drop by drop until the pH of the solution is neutral (somewhere between 20-30 drops). Fill a filter funnel halfway with the product and rinse the sediments with water. To get the product as dry as possible, press it using filter paper. The crystals are allowed to dry naturally. Take the crude product's yield and melting point.
Part D: Procedure for TLC & Column Chromatography
Thin-layer chromatography will be used to determine the distribution of products in your acetylation reaction.
Place a few (5 to 10) drops of methylene chloride, CH2Cl2, in two tiny vials to make solutions of your sublimed ferrocene and dried acetylation product. In one vial, put a little amount of ferrocene (one spatula-tip full) and the acetylation result in the other. After application, the solution should be concentrated enough to leave a dark mark on the TLC plate. If spots on the TLC plate are not visible, add more sample to the solutions and try again. (You can use the same TLC plate as before; simply add the highly concentrated solution directly on top of the first sample.)
The purpose of this part of the experiment is to find the appropriate hexane to ethyl acetate ratio for separating the different ferrocene compounds. The separation of products as a function of solvent polarity is the principle used here.
Begin by separating the products with a pure solvent (hexane or ethyl acetate). After you've gotten those results, choose whole number solvent combination ratios and test them in the same way (i.e. 1:4 hexane to ethyl acetate). TLC plates can be bought at the pharmacy, and micro-capillary tubes will be used as applicators. As illustrated in the illustration, place a piece of filter paper in a tiny jar with a lid and carefully pour in the solvent. Make sure the solvent level is below the point where the TLC plate is visible. On the TLC plate, draw a line to indicate the beginning solvent, then use the capillary to position the TLC plate just above the solvent line. Place the stained TLC plate into the solvent jar with care and cover with a lid. Allow the plate to develop in the solvent until the product spots are 1 cm from the plate's top. If you keep the plate in the solvent for too long, the spots will concentrate at the top and you won't be able to make any useful observations. In your lab notebook, doodle each chromatogram result and make a note of the solvent ratio you used for that plate. Calculate the ratio to front (Rf) value for each spot after obtaining a TLC plate with good separation (no smearing of spots, no spot overlap, etc.).
After that, you'd use column chromatography to gather your isolated products and determine their purities and yields, using the solvent ratio determined by TLC. Unfortunately, due to time limits in 124L, you will not have enough time to complete this task. However, please describe how you would conduct such an experiment and what you would do to determine the purity and yield of your separated products in your lab report.
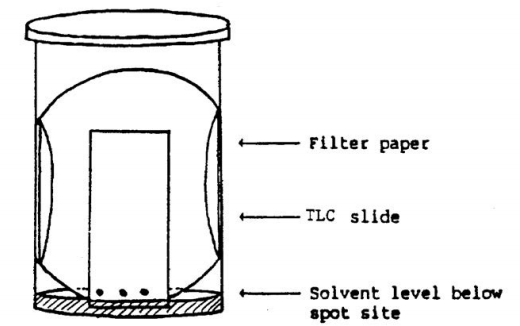
Fig: Apparatus for the separation of Acetylation products using RLC
2.6.1 Aromatic Compounds: Structure:
Because of their distinct scents, benzene-like compounds were previously referred to as aromatic hydrocarbons. An aromatic compound is now defined as any chemical with a benzene ring or some benzene-like characteristics (but not necessarily a strong aroma). The presence of one or more benzene rings in the structure of the aromatic compounds in this text makes them easy to identify. Table lists several example aromatic compounds and their applications, with the benzene ring denoted as C6H5.
Table: Some Representative Aromatic Compounds | ||
Name | Structure | Typical Uses |
Aniline | C6H5–NH2 | Starting material for the synthesis of dyes, drugs, resins, varnishes, perfumes; solvent; vulcanizing rubber |
Benzoic acid | C6H5–COOH | Tobacco curing; food preservation; starting material for the production of colours and other chemical chemicals |
Bromobenzene | C6H5–Br | Solvent; motor oil additive; starting ingredient for the production of several other aromatic compounds |
Nitrobenzene | C6H5–NO2 | Starting material for aniline production; cellulose nitrate solvent; used in soaps and shoe polish |
Phenol | C6H5–OH | a disinfectant; a raw ingredient for the manufacture of resins, medicines, and other chemical compounds. |
Toluene | C6H5–CH3 | Solvent; gasoline octane booster; precursor to benzoic acid, benzaldehyde, and a variety of other chemical compounds |
2.6.2 Polycyclic Aromatic Hydrocarbons
Fused benzene rings—rings that share a common side—make up some common aromatic hydrocarbons. Polycyclic aromatic hydrocarbons are the name for these chemicals (PAHs).
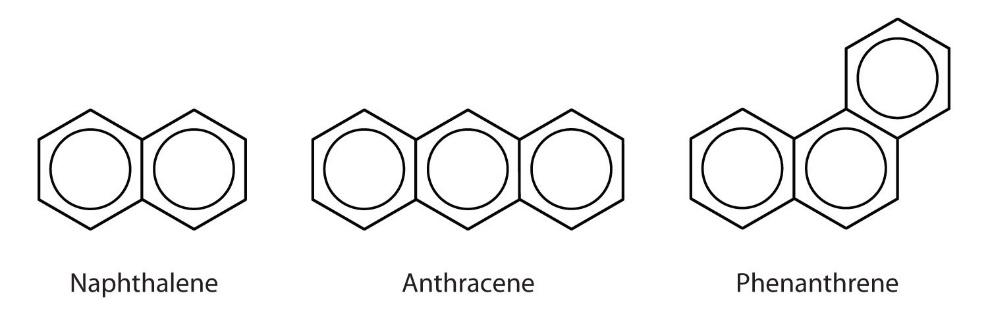
Colorless, crystalline solids, such as the three seen above, are commonly made from coal tar. Naphthalene is utilised in mothballs and has a strong stench. Anthracene is a chemical that is utilised in the production of certain colours. The phenanthrene structure is found in a wide range of naturally occurring compounds, including steroids.
2.6.3 Rules for Aromaticity:
1. Four Key Rules for Aromaticity
There are four characteristics that a molecule must satisfy in order to be aromatic.
It's a case of all or nothing. Aromaticity cannot be achieved if any of these requirements are not met.
- It must first be cyclic.
- Second, each of the ring's atoms must be conjugated.
- Third, the molecule must have [4n+2] pi electrons (we'll go over this in detail later).
- The fourth requirement is that the molecule be planar (usually true if conditions 1-3 are met, but there are rare exceptions)
- Let's take a closer look.
2. Condition #1 for Aromaticity: The Molecule Must Be Cyclic
It's rather simple to tell if a molecule is cyclic. Is there a ring on the finger? If you answered yes, proceed to condition #2. If there isn't a ring, don't bother.
Consider the following example: Although it has the same amount of pi bonds (and pi electrons) as benzene, (Z)-1,3,5 hexatriene is not aromatic. There is no ring, and there is no aromaticity.
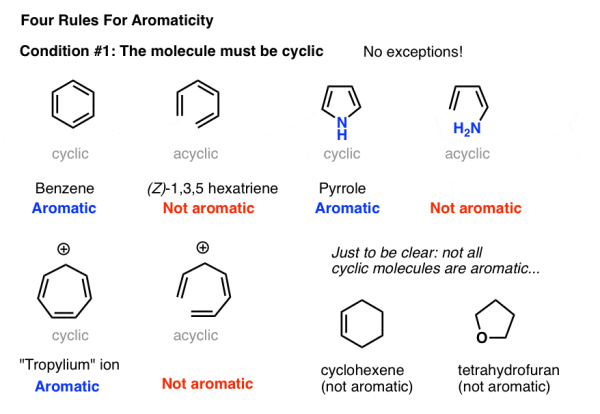
3. Condition #2 For Aromaticity: Every Atom in The Ring Must Be Conjugated
Being cyclic is obviously not a necessary criterion for aromaticity. Take a look at cyclohexene, which is seen to the right. There's nothing fragrant about it.
Aromaticity requires the presence of a continuous ring of p-orbitals surrounding the ring, which builds up into a larger cyclic "pi system."
To put it another way, every atom in the ring must be capable of conjugation with one another.
There are a few different methods to express the same idea.
Alternatively, we can say:
- “There must be an available p orbital for every atom in the ring,” or
- "Every atom in the ring must be able to participate in resonance," says the author.
- Remember that the “available p orbital” criterion applies to atoms with a lone pair, a radical, or an empty p orbital as well as those in a pi bond (e.g. Carbocations). [If you're still not sure, check out this post about conjugation and resonance.]
- An sp3 hybridised atom with four links to atoms is the crucial component that “kills” conjugation. Resonance is impossible for such an atom.
- This is why the pyrrole lone pair (below) isn't as basic as you'd think for a nitrogen. Protonation of the nitrogen breaks the ring's conjugation, resulting in the loss of aromaticity. [Notation 1]
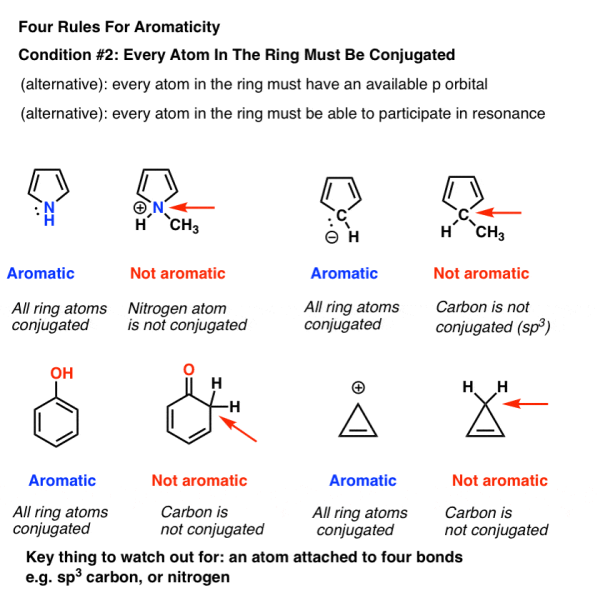
Note: This is an advanced note. Although there are numerous molecules conjugated around the ring's circumference, the inside carbons are sp3 hybridised. It's still considered aromatic.
4. Condition #3 For Aromaticity: The Molecule Must Have [4n+2] Pi Electrons
The cyclic, conjugated molecule must have the correct number of pi electrons as the third criterion. Although both benzene and cyclooctatetraene are cyclic and conjugated, benzene is aromatic while cyclooctatetraene is not. The distinction between benzene and cyclooctatetraene is that benzene has six pi electrons while cyclooctatetraene has eight. [In future posts, we'll explain why this makes a difference.]
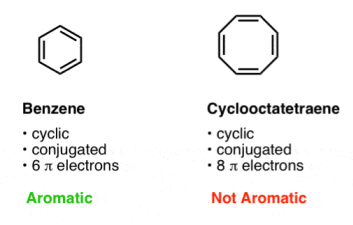
We typically argue that benzene has [4n+2] pi electrons while cyclooctatetraene does not.
However, in organic chemistry, the word [4n+2] causes a lot of misunderstanding. I've had kids stare at a molecule and try to figure out what "n" stands for.
“n” is not a property of the molecule
So for n = 0, we have [4 (0) + 2] = 2
For n = 1, we have [4 (1) + 2] = 6
For n = 2, we have [4 (2) + 2] = 10
For n = 3, we have [4 (3) +2] = 14
And so on.
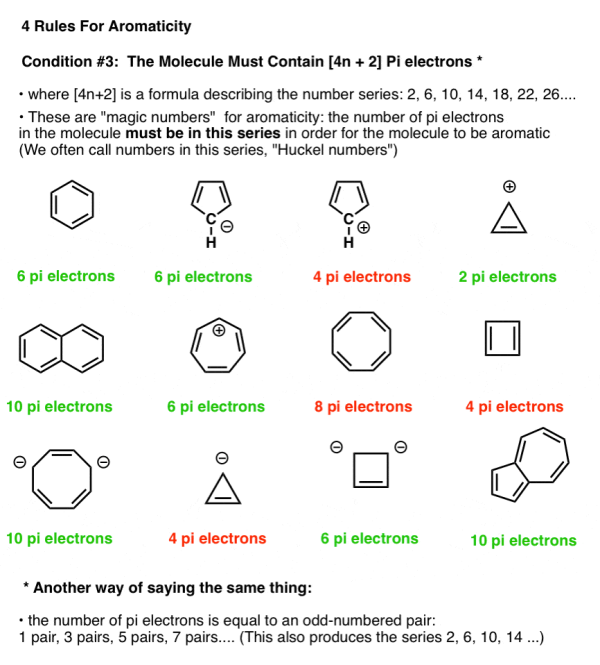
Note that we can count electrons from lone pairs as well as pi bonds (as long as the carbon isn't already involved in a pi bond). As a result, the cyclopentadiene anion has six pi electrons: four from the two double bonds and two from the carbon lone pair.
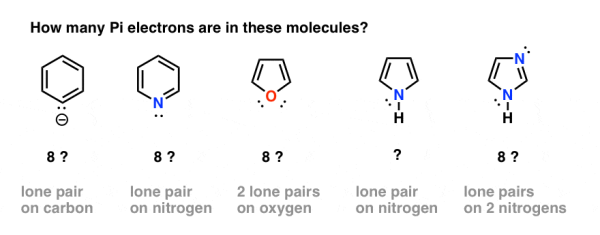
5. Which Electrons Count As “Pi Electrons”, And Which Do Not?
That appears to be simple enough. When we have atoms in the ring that participate in both pi bonding and have a lone pair, however, difficulties can develop. How do we count electrons in the benzene anion (bottom left) or pyridine, for example? Do we count the lone pair electrons as Pi electrons, resulting in an 8-electrons total? Or do we turn a blind eye to them?
It's vital to remember how p orbitals contribute to aromaticity in benzene in order to answer these issues.
Each p orbital in benzene is positioned at a right angle to the ring plane (90°). A single electron resides in each p orbital. By counting the pi bonds, we may determine the total number of pi electrons in benzene: Three pi bonds multiplied by two electrons equals six pi electrons overall.
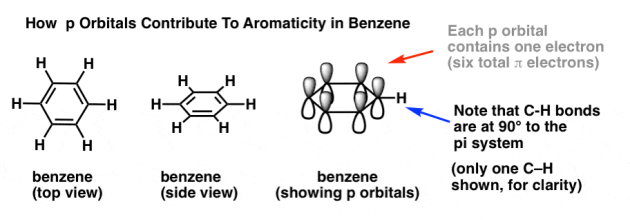
The C-H bonds are at a 90° angle to the pi system.
6. Some Electrons Don’t Count: Pyridine and the Benzene Anion
One of the carbons in the benzene anion possesses a lone pair. Because the p-orbital is involved in the pi system, this lone pair cannot be in a p orbital. Instead, it's in the plane of the ring, at 90 degrees to the pi system.
In other words, because the lone pair on carbon cannot overlap with the pi system, it does not count as a pair of pi electrons.
The lone pair in pyridine is also at right angles to the pi system, as is the case with pyridine.
While you would be tempted to reply they contain 8 pi electrons in each case, the right answer is 6. Both of these compounds are aromatic, and this is a Hückel number.
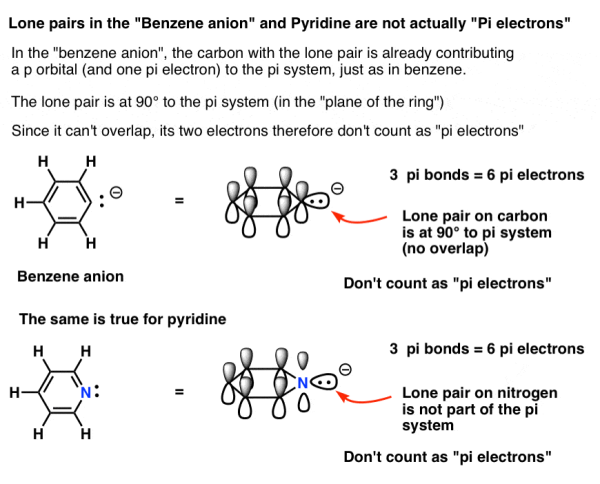
The bottom conclusion is that each ring element can only contribute one p orbital to the pi system.
7. Some Examples Of Aromatic 5-Membered Rings
Some compounds with five-membered rings can be ambiguous as well.
One of the carbons in the cyclopentadiene anion (below) possesses a lone pair. Is it possible for this lone pair to contribute to the Pi system?
The answer is affirmative because the carbon isn't involved in any pi-bonding.
The cyclopentadiene anion has a total of 6 pi electrons: 2 (from the lone pair) + 4 (from the two pi bonds). The cyclopentadiene anion is aromatic, and this is a Hückel number.

Pyrole finds itself in a similar scenario. Although nitrogen has a lone pair, it does not participate in a pi bond (unlike pyridine, above). As a result, it can contribute to the pi system, giving us a total of 6 pi electrons after the four electrons from the two pi bonds are taken into consideration.


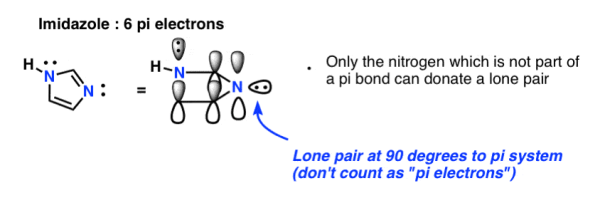
Finally, there's imidazole, a two-nitrogen compound. One nitrogen (the N-H) is not involved in a pi bond and hence can contribute a whole lone pair; the other is, and the lone pair is on the ring plane. Once the two pi bonds are taken into consideration, we have a total of 6 pi electrons.
8. Condition #4 For Aromaticity: The Molecule Must Be Flat
The molecule must also be flat, which is the fourth criterion for aromaticity (planar).
Aromaticity is such a stabilising characteristic (valued at 20-36 kcal/mol) that most cyclic molecules have it.
- Conjugation
- Consists of [4n+2] pi electrons
Will be flat as well. If you give a molecule a large enough potential energy well, it will ultimately fall into it.
It's like the punch line to the old (crude) joke about why dogs acquire a particular energetically advantageous conformation: "Because they can."
The only thing that prevents a molecule that meets the first three characteristics from being flat, as with certain vertebrates, is if the flat conformation is extremely stressed. The chemical known as [10]-annulene, of which an isomer is depicted below left, is one example in this category. The molecule is cyclic, conjugated, and possesses 10 pi electrons in the trans, cis, trans, cis, cis isomer, however the two indicated hydrogens collide while attempting to adopt a flat shape.
Because this punitive Van Der Waals strain prevents the molecule from adopting planarity, it is not aromatic.
The tension is alleviated and the pi bonds can adopt a planar shape when the hydrogens are removed and replaced by a bridging CH2 group. The molecule to the right demonstrates the qualities that an aromatic molecule should have.
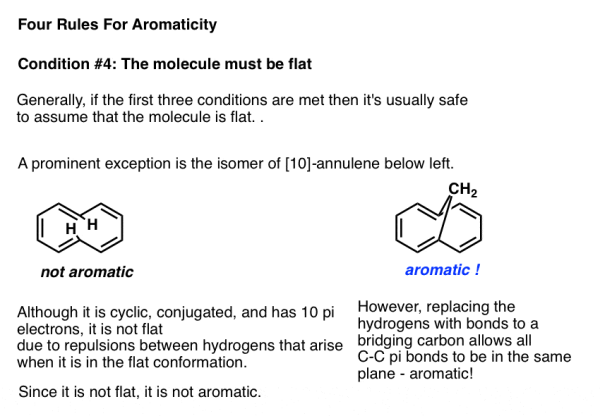
Key Takeaway
- Aromatic rings (also known as aromatic compounds or arenes) are hydrocarbons that contain benzene or a ring structure similar to it. Benzene, C6H6, is commonly shown as a six-carbon ring with alternating double and single bonds: However, there are significant problems with this simple image.
- Alkyl groups are designated using the alkane series convention, which ends in -yl: methyl (one carbon), ethyl (two carbons), propyl (three carbons), and so on. If there are more than six carbons in the substituent, the alkane component is called first, and the aromatic ring component is appended as a suffix.
2.7.1 Aromatic Substitution Reactions:
Substitution Reactions of Benzene and Other Aromatic Compounds
An earlier section described the extraordinary durability of the unsaturated hydrocarbon benzene. The chemical reactivity of benzene differs from that of alkenes in that substitution reactions take precedence over addition reactions, as indicated in the diagram below (some comparable reactions of cyclohexene are shown in the green box).
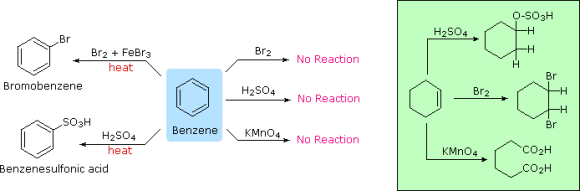
There have been many more benzene substitution reactions discovered; the five most useful are given here (chlorination and bromination are the most common halogenation reactions). Electrophilic Aromatic Substitution is the name given to these reactions because the reagents and conditions used in them are electrophilic. The catalysts and co-reagents are used to produce the strong electrophilic species that are required to complete the first phase of the substitution. In the right hand column, the specific electrophile thought to be involved in each sort of reaction is specified.
Reaction Type | Typical Equation | Electrophile E(+) | |||
Halogenation: | C6H6 | + Cl2 & heat | ——> | C6H5Cl + HCl | Cl(+) or Br(+) |
Nitration: | C6H6 | + HNO3 & heat | ——> | C6H5NO2 + H2O | NO2(+) |
Sulfonation: | C6H6 | + H2SO4 + SO3 | ——> | C6H5SO3H + H2O | SO3H(+) |
Alkylation: | C6H6 | + R-Cl & heat | ——> | C6H5-R + HCl | R(+) |
Acylation: | C6H6 | + RCOCl & heat | ——> | C6H5COR + HCl | RCO(+) |
1. A Mechanism for Electrophilic Substitution Reactions of Benzene
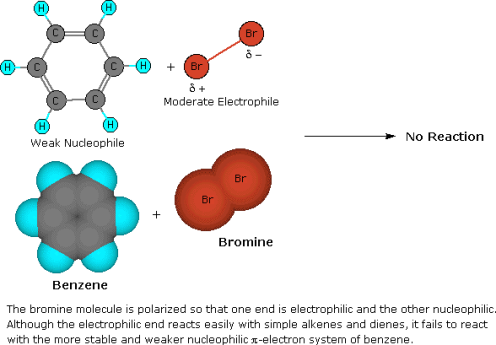
For these electrophilic substitution reactions, a two-step mechanism has been proposed. In the first, slow or rate-determining, step the electrophile forms a sigma-bond to the benzene ring, generating a positively charged benzenonium intermediate. A proton is removed from this intermediate in the second, quick step, yielding a substituted benzene ring. This mechanism for the bromination reaction is depicted in the four-part diagram below. There's also an animated diagram to look at.
Bromination of Benzene - An Example of Electrophilic Aromatic Substitution
This method for electrophilic aromatic substitution should be viewed in the context of other carbocation intermediate-based methods. Alkyl halide SN1 and E1 reactions, as well as alkene Brnsted acid addition reactions, are examples.
To summarize, when carbocation intermediates are formed one can expect them to react further by one or more of the following modes:
1. The cation can form a substitution or addition product by bonding to a nucleophile.
2. A proton may be transferred from the cation to the base, resulting in a double bond product.
3. The cation may rearrange into a more stable carbocation, after which it will react in mode #1 or #2.
The first two forms of reaction are represented by the SN1 and E1 reactions, respectively. The first mode is used in the second phase of alkene addition reactions, and any of these three processes might result in molecular rearrangement if an initial unstable carbocation is generated. Charge delocalization (resonance) stabilises the carbocation intermediate in electrophilic aromatic substitution (the benzenonium ion), preventing rearrangement. In theory, it could react in either mode 1 or 2, but the energy benefit of recreating an aromatic ring makes mode 2 the only option (ie. Proton loss).
2. Substitution Reactions of Benzene Derivatives
When substituted benzene compounds go through electrophilic substitution processes like the ones described above, there are two things to keep in mind:
The first is the compound's relative reactivity in comparison to benzene. Experiments have demonstrated that the substituents on a benzene ring have a significant impact on reactivity. A hydroxy or methoxy substituent, for example, boosts the rate of electrophilic substitution by ten thousand times, as seen in the virtual demonstration with anisole (above). A nitro substituent, on the other hand, reduces the ring's reactivity by a factor of a million. The electron donating or electron withdrawing influence of the substituents, as measured by molecular dipole moments, may be associated with the activation or deactivation of the benzene ring toward electrophilic substitution. The benzene ring is activated for electrophilic attack by electron giving substituents (blue dipoles), while electron withdrawing substituents (red dipoles) deactivate the ring (make it less reactive to electrophilic attack).

The interaction of two effects can explain the influence of a substituent on the reactivity of a benzene ring:
The first is the substituent's inductive impact. Except for metals and carbon, most elements have a far higher electronegativity than hydrogen. As a result, substituents containing nitrogen, oxygen, or halogen atoms form sigma bonds with the aromatic ring, causing inductive electron removal and ring deactivation (left-hand diagram below).
The second impact occurs when a substituent function is conjugated to an aromatic ring. In contrast to the inductive shift, this conjugative interaction permits electron pair donation or withdrawal to or from the benzene ring. Electrons may flow into the aromatic ring via p-conjugation (resonance) if the atom connected to the ring has one or more non-bonding valence shell electron pairs, as do nitrogen, oxygen, and the halogens. Finally, as seen in the right-hand picture, polar double and triple bonds conjugated to the benzene ring can remove electrons. It's worth noting that none of the donors are shown in the resonance examples. The charge distribution in the benzene ring is largest ortho and para to the substituent in both circumstances.
The electron donation by resonance dominates the inductive effect in the case of the nitrogen and oxygen activating groups shown in the top row of the previous diagram, and these compounds have outstanding reactivity in electrophilic substitution processes. Despite the fact that halogen atoms have non-bonding valence electron pairs that participate in p-conjugation, their strong inductive impact dominates, making molecules like chlorobenzene less reactive than benzene. The three examples on the left of the bottom row (in the same diagram) show electron removal via conjugation to polar double or triple bonds, and the inductive effect amplifies the deactivation of the benzene ring in these circumstances. In the same way that alkyl substituents like methyl increase the nucleophilicity of double bonds, they also increase the nucleophilicity of aromatic rings.
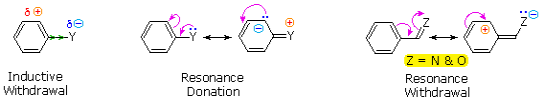
The position at which electrophilic substitution occurs is the second factor that becomes essential in substituted benzene reactions. Because a mono-substituted benzene ring has two equivalent ortho-sites, two equivalent meta-sites, and a single para-site, it can create three different constitutional isomers. The estimated statistical combination of isomeric products would be 40% ortho, 40% meta, and 20% para if reaction happens equally well at all accessible sites. Once again, the type of the substituent has a significant impact on the product ratio. Bromination of methoxybenzene (anisole) occurs quickly and produces primarily the para-bromo isomer, with 10% of the ortho-isomer and barely a trace of the meta-isomer remaining. Bromination of nitrobenzene necessitates high temperatures and yields the meta-bromo isomer as the primary result.

The table below contains some other examples of product isomer distribution in different electrophilic substitutions. It's worth noting that the reaction conditions for these substitution reactions aren't identical, and they must be tweaked to meet the reactivity of the reactant C6H5-Y. Because of anisole's strong reactivity, the first two reactions must be carried out at extremely moderate circumstances (low temperature and little or no catalyst). Because the nitrobenzene reactant in the third example is so unreactive, the reaction must be carried out under extremely severe circumstances.
Y in C6H5–Y | Reaction | % Ortho-Product | % Meta-Product | % Para-Product |
–O–CH3 | Nitration | 30–40 | 0–2 | 60–70 |
–O–CH3 | F-C Acylation | 5–10 | 0–5 | 90–95 |
–NO2 | Nitration | 5–8 | 90–95 | 0–5 |
–CH3 | Nitration | 55–65 | 1–5 | 35–45 |
–CH3 | Sulfonation | 30–35 | 5–10 | 60–65 |
–CH3 | F-C Acylation | 10–15 | 2–8 | 85–90 |
–Br | Nitration | 35–45 | 0–4 | 55–65 |
–Br | Chlorination | 40–45 | 5–10 | 50–60 |
Chemists have developed an empirical taxonomy of the numerous substituent groups often observed in aromatic substitution processes as a result of these and other discoveries. As a result, substituents that engage the benzene ring to electrophilic attack tend to guide substitution to the ortho and para positions. Deactivating substituents, with a few exceptions, such as halogens, direct substitution to the meta location. This classification is summarised in the table below.
Orientation and Reactivity Effects of Ring Substituents | |||||||
Activating Substituents |
| Deactivating Substituents |
| Deactivating Substituents | |||
–O(–) | –NH2 |
| –NO2 | –CO2H |
|
| –F |
Although the information given in the above table is quite valuable for understanding and predicting the course of aromatic substitution reactions, most chemists find it beneficial to grasp the physical factors that contribute to this empirical classification. Inductive and resonance effects have already been utilised to examine the activating or deactivating qualities of substituents, and these same characteristics can be utilised to interpret their influence on substitution orientation.
The first thing to remember is that the proportions of ortho, meta, and para substitution represent the relative rates of substitution at each of these locations in a given example. We can assign the rate of reaction at one of the carbons to be 1.0 if we use the nitration of benzene as a reference. The overall rate would be 6.0 because benzene has six equivalent carbons. We can assign relative rates to the ortho, meta, and para sites in each of these compounds if we investigate the nitration of toluene, tert-butylbenzene, chlorobenzene, and ethyl benzoate in the same way. The overall rate indicated below each structure reflects the 2 to 1 ratio of ortho and meta sites to the para location, as seen in the following picture (coloured red). Divide by six to get the overall relative reaction rates, which are related to benzene as 1.0. In the nitration reaction, the alkyl substituents clearly activate the benzene ring, whereas the chlorine and ester substituents deactivate it.
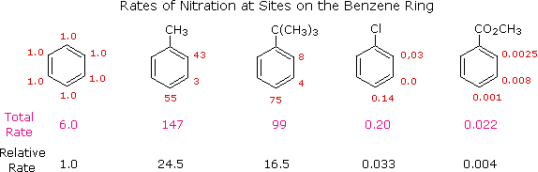
Calculating the proportions of the three substitution isomers from rate data of this type is a simple task. 58.5 percent ortho-nitrotoluene, 37 percent para-nitrotoluene, and only 4.5 percent meta-nitrotoluene are produced by toluene. The greater bulk of the tert-butyl group prevents assault on ortho-sites, with the overall product mixture consisting of 16 percent ortho, 8% meta, and 75% para-nitro. Despite the fact that chlorobenzene is far less reactive than benzene, the rate of ortho and para-substitution far outweighs the rate of meta-substitution, resulting in a product that is 30% ortho and 70% para-nitrochlorobenzene. Finally, as revealed by the relative rates, the benzoic ester produced largely the meta-nitro product (73%) followed by the ortho (22%) and para (5%) isomers. For other substitution processes, equivalent rate and product investigations yield comparable results. Electrophilic chlorination of toluene, for example, is hundreds of times faster than chlorination of benzene, but the relative rates are such that the products are 60% ortho-chlorotoluene, 39% para, and 1% meta-chlorotoluene, a ratio comparable to nitration.
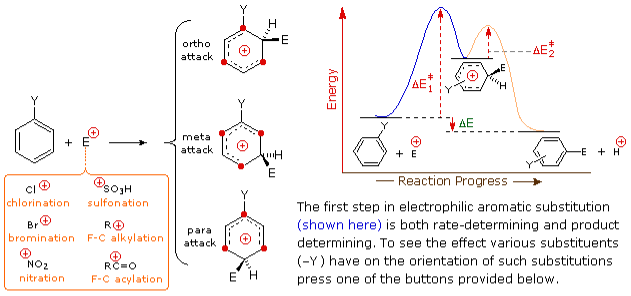
The following interactive diagram depicts how distinct substituents influence the orientation of electrophilic substitution of a benzene ring. The product-determining phase in the substitution mechanism is the first step, which is also the slow or rate-determining step, as shown in the opening picture. It's hardly unexpected, then, that there's a correlation between a substituent's rate-enhancing effect and its site-directing influence. The interactions of a specific substituent with the delocalized positive charge on the benzenonium intermediates created by bonding to the electrophile at each of the three substitution sites are the best way to see the exact effects of that substituent. Using the selection buttons beneath the diagram, you can accomplish this for seven representative substituents.
 | |||||||
|
|
|
|
|
|
|
|
Charge stabilisation is highest for alkyl substituents when they are bound to one of the positively charged carbons of the benzenonium intermediate. This occurs solely in the case of ortho and para electrophilic attack, therefore substituents that enhance the creation of those products are beneficial. Surprisingly, primary alkyl substituents, particularly methyl, provide stronger charge stabilisation than highly substituted groups (note the greater reactivity of toluene compared with tert-butylbenzene).
The atom linked to the aromatic ring has a full or partial positive charge on the nitro (NO2), sulfonic acid (SO3H), and carbonyl (C=O) substituents. Charge repulsion destabilises structures with like-charges close together, hence these substituents hinder ortho and para substitution more than meta substitution. As a result, when electrophilic substitution is pushed, meta-products predominate.
Due to the strong electronegativity of the substituent atoms, the halogen ( X ), OR, and NR2 substituents all have a destabilising inductive impact on a neighbouring positive charge. This would prefer meta-substitution on its own, but these substituent atoms all have non-bonding valence electron pairs that assist to stabilise a neighbouring positive charge by pi-bonding, resulting in charge delocalization. As a result, all of these substituents cause ortho and para sites to be substituted. The balance between inductive electron removal and p-conjugation is such that nitrogen and oxygen substituents have a general stabilising effect on the benzenonium intermediate and significantly boost the rate of substitution, whereas halogen substituents have a general destabilising effect.
3. Characteristics of Specific Substitution Reactions
|
The table on the right repeats the conditions usually used for the aromatic substitution reactions mentioned here. These reagents have varying electrophilic reactivity. For example, we discovered that nitrobenzene nitration happens smoothly at 95oC, yielding meta-dinitrobenzene, whereas nitrobenzene bromination (ferric catalyst) requires a temperature of 140oC. Also, as previously stated, toluene nitrates around 25 times quicker than benzene, whereas toluene chlorinates approximately 500 times quicker than benzene. We can deduce that the nitration reagent is more reactive and less selective than the halogenation reagents based on this.
Water is produced as a by-product of both sulfonation and nitration. The nitration reaction remains unaffected (notice the presence of sulfuric acid as a dehydrating agent), but sulfonation is reversible and is accelerated by the addition of sulphur trioxide, which transforms the water to sulfuric acid. The reversibility of the sulfonation process can be used to remove this functional group on occasion.
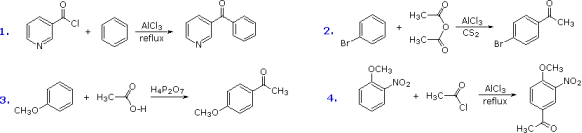
Friedel-Crafts acylation reagents are usually made up of an acyl halide or anhydride and a Lewis acid catalyst like AlCl3. This results in the formation of an acylium cation, R-CO(+), or a similar species. Because such electrophiles aren't particularly reactive, acylation is usually limited to aromatic systems that are at least as reactive as chlorobenzene. Because it is non-reactive and easily removed from the product, carbon disulfide is frequently employed as a solvent. If the substrate is a highly reactive benzene derivative like anisole, the acylating electrophile could come from carboxylic esters or acids. The figure below shows some examples of Friedel-Crafts acylation reactions. The first highlights the possibility of using uncommon acylating chemicals as reactants. The second uses an anhydride acylating reagent, while the third shows how easily anisole reacts, as previously mentioned. The H4P2O7 reagent utilised here is pyrophosphoric acid, a phosphoric acid anhydride. Friedel-Crafts acylation of nitrobenzene does not occur under any circumstances because the nitro group is a strong deactivating substituent. The presence of a second strongly-activating substituent group, on the other hand, allows acylation; the reaction site is preferred by both substituents.
A common characteristic of the halogenation, nitration, sulfonation and acylation
The introduction of a deactivating substituent on the benzene ring is a key feature of these reactions. As a result, we rarely have to be concerned about disubstitution products forming. Friedel-Crafts alkylation, on the other hand, involves the addition of an activating substituent (an alkyl group), allowing for many substitutions. When benzene is alkylated, as in the following synthesis of tert-butylbenzene, a substantial excess of this reactant favours the mono-alkylated product. When the molar ratio of benzene to alkyl halide is less than 1:1, the main result is para-ditert-butylbenzene.
C6H6 (large excess) + (CH3)3C-Cl + AlCl3 ——> C6H5-C(CH3)3 + HCl
Alkyl halides (as mentioned above), alkenes + strong acid, and alcohols + strong acid can all be used to make the carbocation electrophiles needed for alkylation. Because 1o-carbocations are prone to rearrangement, Friedel-Crafts alkylation is rarely used to introduce 1o-alkyl substituents greater than ethyl. Excess benzene, for example, can be converted to isopropyl benzene by reacting it with 1-chloropropane and aluminium chloride (cumene).
——> C6H5-CH(CH3)2 + HCl ——> C6H6 (large excess) + CH3CH2CH2-Cl + AlCl3 ——> C6H5-CH(CH3)2 + HCl
Additional Friedel-Crafts alkylation processes are depicted in the diagram below.
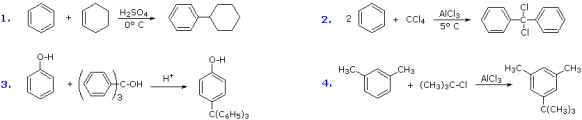
Alkenes and alcohols can be used as sources of the electrophilic carbocation reactant in the first and third cases. Due to significant resonance charge delocalization, the triphenylmethyl cation formed in the third scenario is very unreactive and only substitutes highly activated aromatic rings. The second example demonstrates an unusual instance in which the alkylating agent is a polychloro reactant. To avoid tri-alkylation of this reagent, which is slowed by steric hindrance, a four-fold excess of carbon tetrachloride is utilised. The fourth example shows how alkylation reactions of activated benzene rings frequently have poor orientational selectivity. At the least inhibited location, the bulky tert-butyl group is linked to the reactive meta-xylene ring. Because polyalkyl benzenes rearrange under Friedel-Crafts conditions, this may not be the site of initial bonding (para-dipropyl benzene rearranges to meta-dipropyl benzene on heating with AlCl3).
Separation of isomer mixtures is a practical concern when using electrophilic aromatic substitution processes in synthesis. This is especially true in ortho-para substitutions, which frequently result in large amounts of the minor isomer. Except for specific reactions of toluene and similar alkyl benzenes, para-isomers usually win out. The fact that para-isomers have much higher melting temperatures than their ortho counterparts aids separation of these combinations; as a result, fractional crystallisation is typically an efficient isolation procedure. Separation of trace isomers is usually not an issue because meta-substitution favours a single product.
References:
1. Das Asim K., Fundamentals of Inorganic Chemistry, Vol. II, CBS Publications, 2nd Ed. 2010.
2. Selected Topic in Inorganic Chemistry, Mallick, Madan and Tuli, S. Chand Publisher. 17th Ed. 2010.
3. Mehrotra R.C. And Singh, A. Organometallic Chemistry, New Age International Publishers, 2nd Edn, 2000.
4. Gupta B. D. And Elias A. J., Basic Organometallic Chemistry, 2nd Edn., University Press (2013).




