Unit - 3
Catalysis by Organometallic Compounds
3.1.1 Catalytic Applications of Organometallic Compounds- Alkene Metathesis
With the rapid discovery of new catalysts in this field, the use of organometallic chemistry in homogeneous catalysis is steadily increasing. Organometallic catalysis' advantages have now spread throughout the chemical world, from industry to small-scale organic synthesis in academic laboratories. Several of these organometallic complexes in homogeneous catalysis applications have left an indelible mark on the ongoing developmental process that is constantly transforming our daily lives. Alkene metathesis, which is described in this chapter, is an example of such a success story.
3.1.2 Alkene metathesis
The use of alkene metathesis reactions to make unsaturated olefinic compounds and their polymeric counterparts is gaining popularity. Depending on the reaction conditions of the metathesis reaction, a metal carbene intermediate reacts with olefins to produce various olefinic compounds or even unsaturated olefinic polymers. Metathesis is an unusual reaction in which a C=C bond is broken and a new unsaturated olefin is formed during catalysis.

Only a few metalcarbene catalysts have been found to be functional group tolerant, despite the fact that a large number of metalcarbene catalysts have been developed for the metathesis reaction. As a result, developing functional group tolerant catalysts has been a crucial step in broadening the utility of the metathesis reaction. Early-transition metal based carbene catalysts, such as those based on Ti, are highly oxophilic and thus intolerant of functional groups in this regard. The more electron-rich Mo and W-based catalysts, on the other hand, are classified as intermediate. Finally, Ru catalysts based on late-transition metals are found to be extremely tolerant of functional groups while still exhibiting high reactivity toward olefinic bonds. The Grubb's Ru catalyst, which is simple to use, and the Schrock's Mo catalyst, which has a high activity, are noteworthy in this context.
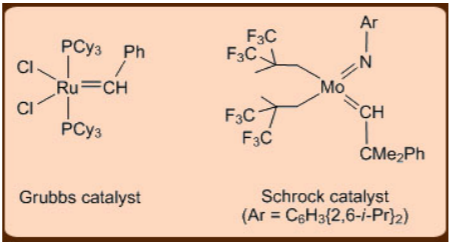
Figure: The Grubb’s and the Schrock’s catalyst.
The term "metathesis" refers to a group of reactions that all involve the "cutting and stitching" of olefinic bonds to produce various unsaturated products. Because the olefinic ends are exchanged when two different olefin substrates are used, this reaction is known as "cross metathesis."

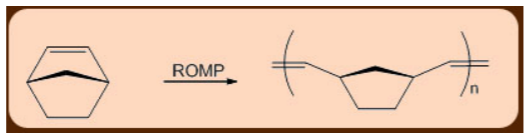
In systems where the ring strain in the final product is not too high, the metathesis reactions can even extend to conjugated dienes, which can undergo Ring Closing Metathesis (RCM). Ring Opening Metathesis (ROM) is the reversal of Ring Closing Metathesis (RCM), and it's usually preferred when there's a lot of C2H4.
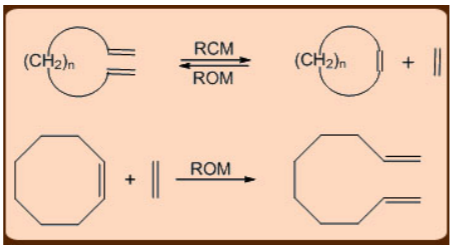
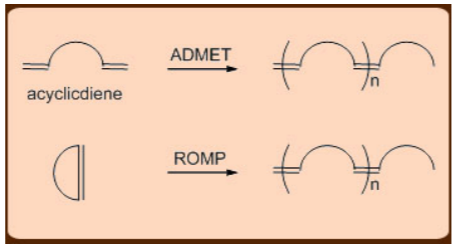
The Acyclic Diene Metathesis (ADMET) and the Ring Opening Metathesis Polymerization (ROMP), in which the relief of cycloalkene ring-strain drives the polymerization reaction forward, are two common metathesis variants used in the production of polymers. Both of these reactions produce living long chain polymers, making them useful for making block copolymers (AAABBBB) n.
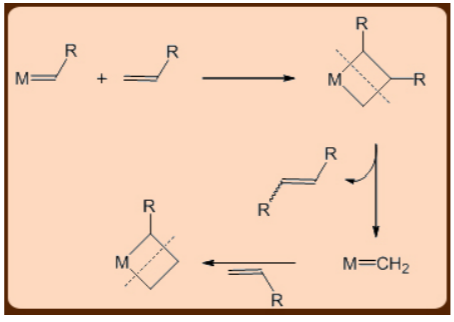
The mechanism of the metathesis reaction has been debated, but the one involving a metalacyclobutane intermediate has gained traction.
The metathesis reaction has resulted in a number of important industrial applications, such as the commercial synthesis of housefly pheromone.
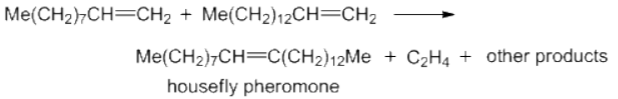
Similarly, because of its exceptional strength owing to its cross-linked nature, the polycyclopentadiene polymer, which is formed from the Ring Opening Metathesis Polymerization (ROMP) of dicyclopentadiene substrate, is used in bullet proof applications.
3.1.3 Designing organometallic compounds for catalysis.
Bioorganometallic chemistry is an area of research that is rapidly evolving. Organometallic compounds have recently provided a rich platform for the development of efficient catalysts for olefin metathesis and transfer hydrogenation, for example. Thermodynamics and kinetics of ligand substitution and redox reactions of metal ions, particularly RuII, are controlled using electronic and steric effects. Is it possible to design targeted organometallic drugs that incorporate similar features? Through the incorporation of outer-sphere recognition of targets and controlled activation features based on ligand substitution as well as metal- and ligand-based redox processes, such complexes have the potential to provide novel drug action mechanisms. The 6-arene, 5-cyclopentadienyl sandwich, and half-sandwich complexes of FeII, RuII, OsII, and IrIII with promising activity against cancer, malaria, and other conditions are the focus of this paper.
Catalysis is an important point to remember. Catalysis frequently involves organometallic complexes. Hydrogenation, hydrosilylation, hydrocyanation, olefin metathesis, alkene polymerization, alkene oligomerization, hydrocarboxylation, methanol carbonylation, and hydroformylation are some of the most commonly used industrial processes.
Homogeneous catalysts, heterogeneous catalysts, and biocatalysts are the three types of catalysts and catalytic reactions (usually called enzymes).
Wilkinson’s Catalyst- Hydrogenation of Olefins (Alkenes)- Mechanism
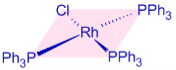
RhCl(PPh3)3 – Wilkinson's catalyst is chlortris(triphenylphosphine)rhodium(I). It is employed as a catalyst for homogeneous hydrogenation. It's a 16-electron diamagnetic square planar complex. Wilkinson's catalyst has a +1 oxidation state for Rhodium and demonstrates dsp2 hybridization for Rhodium.
Preparation:
* It can be prepared by reacting RhCl3.3H2O with excess PPh3 in EtOH.

Uses:
It is used in the selective hydrogenation of alkenes and alkynes without affecting the functional groups like: C=O, CN, NO2, Aryl, CO2R etc.
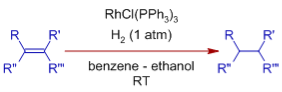
Mechanism of Hydrogenation - Catalytic Cycle
Wilkinson's catalyst is a pre-catalyst that is transformed to an active state before entering the catalytic cycle by removing one triphenylphosphine ligand. In most cases, the solvent molecule fills the empty space.
Initially, the catalyst uses an oxidative addition process to activate the molecular dihydrogen, resulting in an 18 valence electron dihydrido complex. Rh's oxidation state has been raised to +3. In the next stage, the generated dihydrido complex attaches to the olefin, resulting in the loss of solvent or PPh3 ligand. This method is known as the dihydride path because the activation of dihydrogen occurs before the addition of olefin.
At the double bond, one of the hydrogens undergoes migratory insertion. This is a slow step, referred to as the Rate Determining Step (RDS).
An irreversible reductive elimination phase that completes the catalytic cycle releases the alkane quickly and completely. The catalyst is renewed once the oxidation state of Rh is reduced to +1.
However, under the given reaction circumstances, additional pathways and intermediates are also feasible (see below).
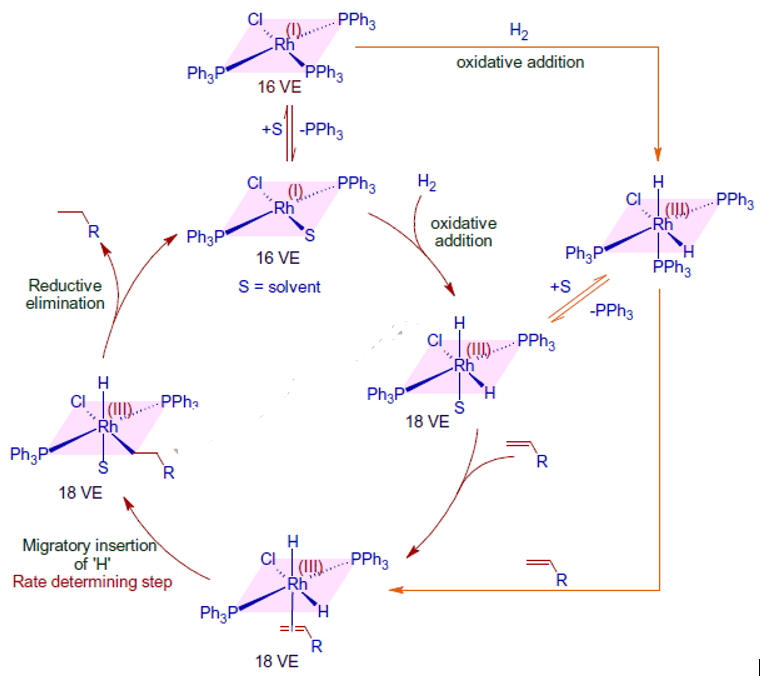
The following observations back up the above mechanism.
* When too much PPh3 is introduced, the rate of reaction slows down, indicating that one of the PPh3 ligands dissociates before dihydrogen activation. Strong -acids, such as ethylene, are thought to behave as poisons because they bind tightly to the electron-rich Rh metal core and prevent hydrogenation.
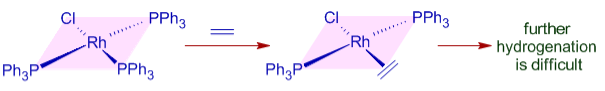
Despite the fact that ethylene cannot be hydrogenated in the presence of Wilkinson's catalyst under normal conditions, hydrogen transfer can be accomplished with a premade dihydrido complex.
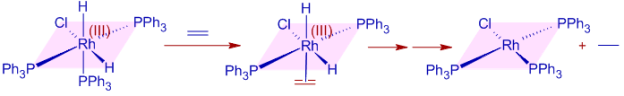
As the alkyl group substitution on the double bond increases, the rate of hydrogenation decreases, mirroring their relative binding affinities to the metal centre. Part of it is related to steric considerations.
As a result, oxidative addition of dihydrogen prior to olefin binding is critical for effective hydrogenation. When an equimolar mixture of H2 and D2 is employed, there is negligible scrambling of H/D in the result.
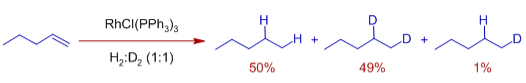
This suggests the development of a dihydrido complex that transfers both of its hydrido ligands to the olefin, as well as the speed and irreversibility of the final reductive elimination phase.
Selective Hydrogenation by Wilkinson's Catalyst
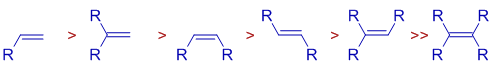
Selective hydrogenations can be achieved using Wilkinson's catalyst. Double bonds that are less substituted and sterically less hindered are selectively hydrogenated.
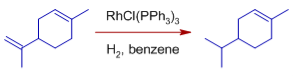
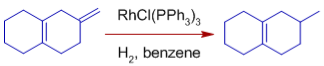
* Exocyclic double bonds are hydrogenated more selectively than endocyclic double bonds.
* Cis alkenes decompose more quickly than trans alkenes.
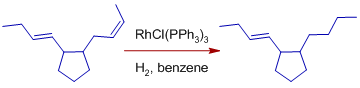
Over conjugated dienes, isolated double bonds are rapidly hydrogenated. Over conjugated dienes, isolated double bonds are rapidly hydrogenated.

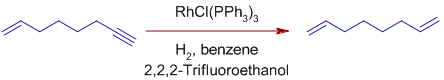
* Terminal alkynes and terminal alkenes are hydrogenated at a faster rate than terminal alkenes. Use of acidic alcoholic co-solvents can improve selectivity.
* C=O, C=N, NO2, Aryl, CO2R, and other functional groups are unaffected. Wilkinson's catalyst's compatibility with polar multiple bonds implies that the metal hydride bond is predominantly covalent.
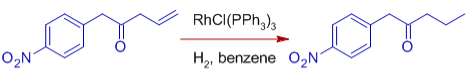
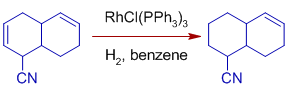
Note that sterically unrestricted aldehyde groups are vulnerable to decarbonylation, making the catalyst useless. Hydrogenation occurs more quickly on unsaturated substrates with polar functionality. It could be due to the olefin's facile coordination to the catalyst, which is aided by the polar functional group.
STEREOCHEMISTRY OF WILKINSON'S HYDROGENATION
Wilkinson's catalyst catalyses stereospecific syn hydrometallation of the multiple bond followed by stereospecific reductive elimination in hydrogenations. As a result, when olefins or alkynes are hydrogenated, they produce syn addition products.
For example, diastereoselective hydrogenation of Maleic acid or Fumaric acid with D2 in the presence of Wilkinson's catalyst. When Maleic acid is hydrogenated with D2, only the meso molecule is produced.
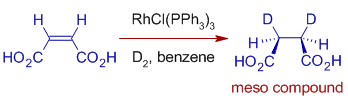
With fumaric acid, however, a racemic mixture is generated. With fumaric acid, however, a racemic mixture is generated.
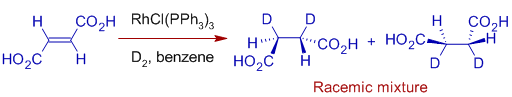
As a result, Wilkinson's hydrogenation is stereospecific and stereoselective.
It is stereospecific in that each stereo isomer produces only one stereo isomer as a unique product. It's owing to the mechanism's stereotypical nature.
It is stereoselective, nevertheless, because only one diastereomer is generated as the primary result. Other mechanisms working under response conditions also contribute to this.
The major result of hydrogenation of alkynes is cis alkenes.
Wilkinson's hydrogenation is diastereoselective, as evidenced by the creation of an endo product as the primary product with the following bicyclic system. The catalyst binds to the double bond on the bicyclic system's least hindered exo face, followed by the syn addition of two hydrogen atoms.
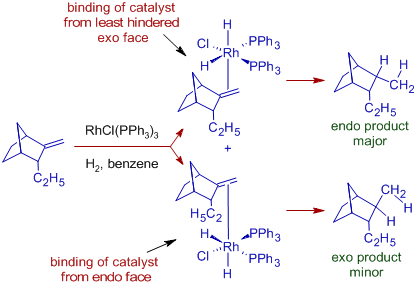
DECARBONYLATION WITH WILKINSON'S CATALYST
Wilkinson's catalyst can be used to decarbonylate aldehydes via the oxidative addition method since Rh(II) is a low valent and coordinately unsaturated metal that can bind to CO strongly.
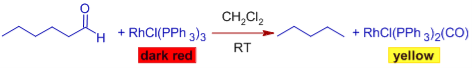
Mechanism:
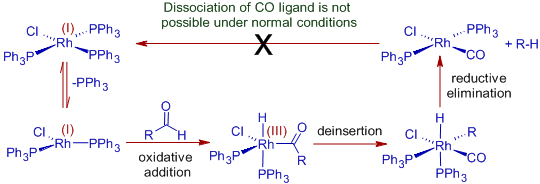
Because the catalyst cannot be replenished, the process is non-catalytic. The complex generated during the process, chlorocarbonylbis(triphenylphosphine) rhodium, is quite stable, and the CO ligand cannot be dissociated at mild temperatures. To carry out this conversion, stoichiometric amounts of the complex are required.
However, using DiPhenylPhosphorylAzide (DPPA), which eliminates the CO ligand from chlorocarbonylbis(triphenylphosphine) rhodium to recreate the active form of catalyst, it is possible to make the decarbonylation reaction catalytic with Wilkinson's catalyst.
Properties of Wilkinson’s Catalyst
Physical Properties
Wilkinson's catalyst has a molecular mass of 925.22 g/mole.
It has a melting point of 518 to 523 degrees Celsius.
Water does not dissolve it. However, it is soluble in benzene and tetrahydrofuran, which are hydrocarbon-based solvents.
Chemical Properties
Wilkinson's catalyst is designed with a square planar coordination geometry.
RhCl(CO)(PPh3)2 is formed when it reacts with carbon monoxide.
When mixed with benzene, this coordination compound dimerizes. [RhCl(PPh3)2] is the chemical formula for the dimer's chemical composition.
Homogeneous Catalysis
"When added to a reaction mixture, a catalyst changes the rate at which the system achieves equilibrium without undergoing a permanent chemical change." (Penguin Dictionary of Chemistry, Sharp, 1981) A catalyst speeds up the time it takes for the system to reach equilibrium, but it doesn't change the equilibrium because it also speeds up the back reaction. It is also necessary to define the following terms:
· A compound or complex that undergoes a reaction to produce the active catalyst.
· An autocatalyst is a reaction product that also serves as a catalyst in the same reaction.
· A compound that slows or stops a reaction is known as an inhibitor.
· TON = moles of product per mole of catalyst; substrate = molecule on which the catalyst acts; percent conversion = percent of the substrate converted to product; turnover number (TON) = moles of product per mole of catalyst (unitless)
· TOF = moles of product per mole of catalyst per hour (units are h-1)
A homogeneous catalyst is one in which the catalyst and the substrate are both in the same phase. An organic molecule, a salt, or a transition metal complex could all serve as homogeneous catalysts. The catalyst does not require a homogeneous solution. Homogeneous hydrogenation, for example, has two phases: hydrogen in the gas phase and a liquid phase containing the catalyst and the substrate (an olefin). The catalyst is a homogeneous catalyst, despite the fact that the system is technically heterogeneous.
Despite the fact that heterogeneous catalysts have long been the industry standard, homogeneous catalysts have a number of advantages (Table 1). Many industrial processes rely on them.
Table. Comparison of homogeneous and heterogeneous catalysis:
Heterogeneous | Homogeneous |
Simple to prepare | Complicated synthesis |
Very stable | Less stable |
No solvent restrictions | Solvent restrictions |
Easy to separate from products, solvents | Difficult to separate |
Difficult to characterize | Simple to characterize (NMR, X-ray) |
Only surface atoms used | All metal atoms used |
Poisons easily | Somewhat more resistant |
Low selectivity | High and tunable selectivity |
Empirical matching of catalyst and substrate | Catalyst design |
A cycle of several steps, each of which can also occur as a stoichiometric reaction, is usually the mechanism of a homogeneously-catalyzed reaction. Oxidative addition, reductive elimination, and insertion are the three most common primary steps in transition metal complexes.
The metal oxidation number increases by two units, the X-Y bond is cleaved, and new M-X and M-Y bonds are formed during the oxidative addition of a molecule X-Y (e.g. H2, HCl, RS-SR, Br2, R-I) to a metal fragment LnM. The metal's electron count increases by two.
As a result, oxidative addition is not possible if the metal already has 18 valence electrons or if it lacks an energetically accessible oxidation number two units higher than the current number.
The opposite of oxidative addition is reductive elimination. The metal oxidation number is decreased by two units, the M-X and M-Y bonds are cleaved, and a new X-Y bond is formed during the reductive elimination of a molecule X-Y from a metal fragment LnM(X)(Y). The metal's electron count drops by two. If the metal does not have an energetically accessible oxidation number two units below the current number, reductive elimination is not possible.
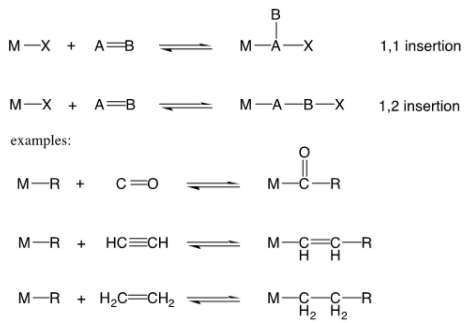
An unsaturated molecule A=B inserts itself into a metal-ligand bond M-X in an insertion reaction (where X is an anionic ligand).
The A-B bond has a one-unit decrease in bond order (i.e. a double bond goes to a single bond, a triple bond goes to a double bond). There are two types of insertion geometries: 1,1 and 1,2. By observing how the molecule binds to the metal as a ligand, you can predict which scenario will occur. When the molecule A=B binds end-on to the metal, 1,1-insertion usually occurs (for example, carbon monoxide). When the molecule A=B binds side-on to the metal, 1,2-insertion usually occurs (for example, an alkene)
A catalytic cycle combines these simple steps (along with a few others). Finding the catalytic cycle for a specific catalytic reaction, on the other hand, can be extremely difficult. A common method is to observe individual steps as stoichiometric reactions and then propose a catalytic cycle as a hypothesis; the mechanism must be consistent with the observed kinetics. Despite this, the mechanism may only match the observations for one substrate, one temperature, and one solvent after all of this work. Changing the substrate, temperature, or solvent may trigger the emergence of a completely new mechanism.
Another issue is that any species detected by spectroscopy during catalysis could actually be a species not involved in the catalytic cycle. If a species can be observed, it is almost certainly too stable to be a part of a fast catalytic cycle.
Homogeneous Hydrogenation
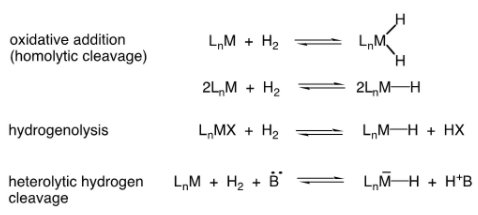
When it comes to homogeneous hydrogenation, hydride ligands are crucial. At room temperature, hydrogen gas (H2) does not react with alkenes. By forming hydride complexes, metal complexes activate hydrogen and transfer it to alkenes. Hydrogenolysis, oxidative addition of H2, or heterolytic activation can all be used to create hydride complexes.
The hydrogen from the metal to the alkene must also be transferred. Hydrogen atom transfer, the hydride pathway, and the unsaturated pathway are the three most common ways for this to happen. Only the order in which hydrogen and alkene bind to the metal differs in the latter two routes.
MH + R2C=CR2 M• + R2HC-CR2• H atom transfer (radical hydr.)
MH2 + R2C=CR2 M(H)2(R2C=CR2) ® M(H)(CR2CHR2) ® M + CHR2CHR2 hydride pathway
M(R2C=CR2) + H2 M(H)2(R2C=CR2) ® M(H)(CR2CHR2) ® M + CHR2CHR2 unsaturated pathway
Wilkinson and Coffey independently discovered chlorotris(triphenylphosphine) rhodium(I), now known as "Wilkinson's catalyst," in 1964. The direct reaction of rhodium chloride with triphenylphosphine is the most straightforward way to make this complex, though it does not work well with other phosphines.
This catalyst was the first to be able to rapidly hydrogenate unconjugated alkenes and alkynes at extremely mild conditions (1 atm of H2 and at room temperature). The hydrogenation is usually performed in benzene with a polar cosolvent like ethanol. The polar solvent might facilitate migratory insertion, which is rate limiting.

Although some olefins, such as ethylene and 1,3 butadiene, are slowly reduced under ambient conditions and can act as powerful competitive inhibitors, steric factors play the most important role in determining olefin reactivity.
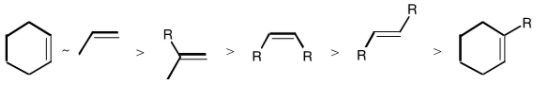
The complex slightly dissociates in solution, and this is thought to be important to the mechanism.
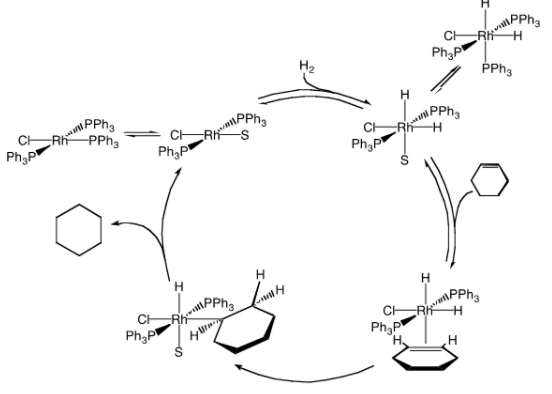
The mechanism for hydrogenation of cyclohexene is shown below (S = solvent), but keep in mind that the mechanism for hydrogenation of styrene is different, and hydrogenation of ethylene involves yet another mechanism.
Wilkinson's catalyst should be referred to as "Wilkinson's catalyst precursor" if this mechanism is correct.
Hydroformylation (Co salts)
Organometallic chemistry is experiencing unprecedented success in homogeneous catalysis, and this is a truly exciting time for the field. In the areas of asymmetric hydrogenation (Nyori and Knowles in 2001), olefin metathesis (Grubbs, Schrock, and Chauvin in 2006), and palladium mediated C-C cross coupling reactions (Grubbs, Schrock, and Chauvin in 2006), the field of organometallic chemistry has grown rapidly, as evidenced by the award of three Nobel prizes in less than a decade (Suzuki, Negishi and Heck, 2010). Below are a few representative examples of such seminal organometallic compound discoveries in homogeneous catalysis.
Hydroformylation reaction
Hydroformylation, also known as the "oxo" process, is a Co or Rh catalysed reaction that produces value-added aldehydes by combining olefins with CO and H2.
Otto Roelen discovered the reaction in 1938, and it quickly grew in scope and scale in terms of aldehyde production around the world. The hydroformylation reaction was catalysed by metal hydride complexes based on rhodium, HRh(CO)(PPh3)3, and cobalt, HCo(CO)4.
Hydroformylation, also known as oxo synthesis or oxo process, is a widely used industrial process for producing aldehydes from alkenes that uses a homogeneously catalysed reaction. 1st. A formyl group (CHO) and a hydrogen atom are introduced into a carbon-carbon double bond in this chemical reaction. Since its inception in 1938, this process has continued to expand: in 1995, the production capacity was 6.6106 tonnes. It's crucial because the aldehydes that result are easily converted into a variety of secondary products. The resulting aldehydes, for example, are hydrogenated to produce alcohols, which are then used to make plasticizers or detergents.Hydroformylation is also used in specialty chemicals like fragrances and natural products. One of the most significant achievements of twentieth-century industrial chemistry is the development of hydroformylation, which originated in the German coal-based industry.
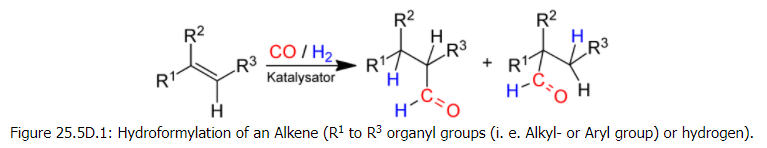
Figure: Hydroformylation of an Alkene (R1 to R3 organyl groups (i. e. Alkyl- or Aryl group) or hydrogen).
Wacker process
The Wacker oxidation is the reaction in which a terminal or 1,2-disubstituted alkene is converted to a ketone using catalytic palladium(II), water, and a co-oxidant. Aldehydes, allylic/vinylic ethers, and allylic/vinylic amines are produced via various reactions. The Wacker oxidation has been widely used in organic synthesis due to the simplicity with which terminal alkenes may be synthesised and the adaptability of the methyl ketone group created by the reaction.
Introduction
The stoichiometric conversion of ethylene to acetaldehyde by an acidic, aqueous solution of PdCl2 was found over a century ago, but it took another fifty years for a catalytic process to be developed. A comparable reaction occurs in an aqueous, acidic solution of catalytic PdCl2 and a stoichiometric quantity of CuCl2 through which oxygen is bubbled, according to Wacker Chemie researchers in 1959.
(1)
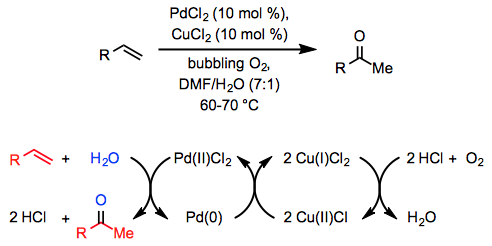
The Wacker technique has been widely used in organic synthesis and has been extended to additional types of substrates and products since its initial report. A co-solvent is usually used in conjunction with water to facilitate mixing of the organic reactants with the aqueous phase. Dimethylformamide (DMF) is a popular choice; the "Tsuji-Wacker oxidation" occurs when DMF is employed as a co-solvent with a stoichiometric quantity of CuCl under balloon pressure of oxygen. The Wacker oxidation is most commonly used in chemical synthesis to install a methyl ketone molecule, which can then be nucleophilically added to or deprotonated to generate an enolate.
Mechanism and Stereochemistry
Prevailing Mechanism: Water Nucleophile
Experimental and theoretical research into the Wacker oxidation mechanism has been conducted. Coordination of the alkene to the palladium centre forms -complex 2 in the first phase of the Wacker oxidation. The relative sluggishness of electron-poor alkenes, which require larger catalyst loadings than unactivated alkenes, serves as evidence for this step. Depending on the method of hydroxypalladation, the result is either zwitterionic complex 3 or neutral complex 4. Hydride elimination then happens, yielding enol complex 5, which re-inserts into the Pd-H bond to yield complex 6. The involvement of chloride-assisted deprotonation in the succeeding step, which yields the product and palladium, is supported by computational studies. Palladium(0) is then oxidised by copper(II), producing palladium(II) species 1 as a result. Copper(IIparticipation )'s in the process is yet unknown.
(2) For the Wacker oxidation, lladation is a critical concern. Hydroxypalladation can take place in a syn or anti fashion, depending on whether an inner-sphere mechanism or a nucleophilic attack on the coordinated alkene is used. Although the stereocenter in 4 is eventually eliminated during elimination, the hydroxypalladation mode can affect the reaction's site selectivity. Water added in a Markovnikov-type reaction to the alkene's more substituted carbon produces a methyl ketone, while water assault on the less substituted carbon produces an aldehyde. The mechanism of hydroxypalladation has been demonstrated to influence the distribution of ketone and aldehyde products, as well as the stereoselectivity of Wacker cyclization reactions.
(3)
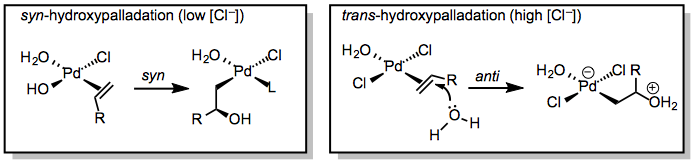
Syn-hydroxypalladation appears to be the norm at low chloride ion concentrations. Water binds to the metal and causes migratory insertion into the Pd-OH bond, resulting in the direct generation of 4. Anti-hydroxypalladation can occur when large quantities of chloride ion compete with hydroxide for binding to palladium.
Prevailing Mechanism: Alcohol Nucleophiles
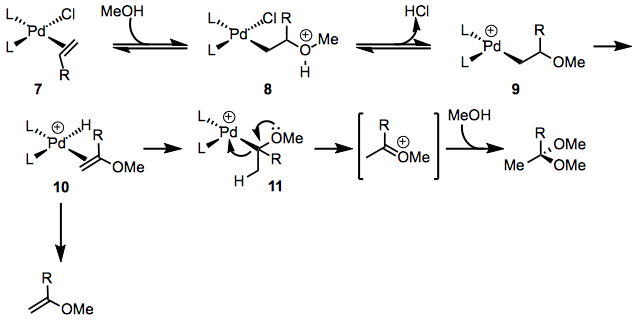
In the Wacker process, alcohols can also be used as nucleophiles. The earliest steps of the mechanism are identical to those of Wacker oxidation with water, but at alkoxy complex 11, the mechanisms diverge. Palladium is removed to produce an oxocarbenium ion, which can be trapped by a solvent to produce an acetal. The intermediacy of complex 10 is supported by small amounts of vinyl ether derivatives.
(4)
Enantioselective Variants
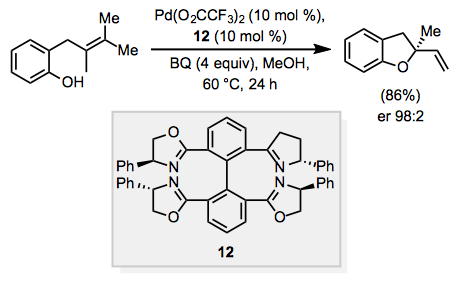
When a tethered nucleophile, such as a hydroxyl group or a protected amine, is present in the substrate, the nucleophile may react with the -hydride to create a cyclic, allylic, or vinylic ether or amine. When the result is allylic, this process is known as "Wacker cyclization," and a stereocenter can be produced. The Wacker cyclization has been made enantioselective using chiral ligands. When tetra(dihydrooxazoline) ligand 12 was combined with benzoquinone (BQ) as a co-oxidant, an allylic ether product with an enantiomeric ratio of 98:2 was formed.
(5)
Through acetal synthesis, the Wacker method can also establish a stereocenter in 1,1-disubstituted alkenes. The use of a chiral auxiliary in the alkene has yielded chiral acetals with a high diastereomeric ratio. The acetal develops at the alkene's more electron-deficient location
(6)
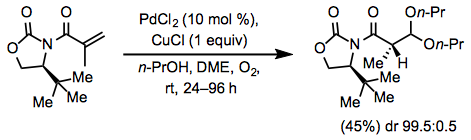
Scope and Limitations
The Wacker approach can also establish a stereocenter in 1,1-disubstituted alkenes using acetal synthesis. With the addition of a chiral auxiliary to the alkene, chiral acetals with a high diastereomeric ratio have been produced. At the alkene's more electron-deficient site, the acetal forms
(7)
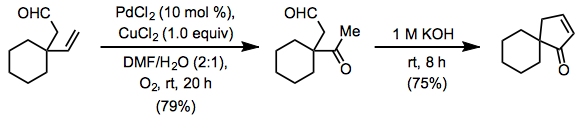
For substrates with acid-sensitive functionality, such as acetals and silyl ethers, the original Wacker procedure and Tsuji-Wacker conditions can cause issues. When copper(II) chloride is used, it produces highly acidic hydrochloric acid. A copper(II) acetate-based approach has been developed to address this problem. Some acid-sensitive organisms can tolerate the gentler acetic acid produced by this procedure A yield of only 56 percent was produced under comparable conditions using copper(I) chloride.
(8)
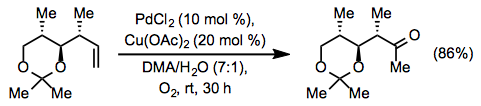
Unprotected amines have a strong palladium coordination and so present complications with Wacker oxidations. Amines containing electron-withdrawing substituents frequently do not interfere with Wacker oxidations, but they may participate in aza-Wacker cyclizations if properly positioned in the substrate (see below).
Only terminal and 1,1-disubstituted alkenes react in the Wacker oxidation; tri-substituted alkenes are entirely inert. Except under very specific circumstances, terminal alkenes are normally oxidised selectively over internal alkenes.
(9)
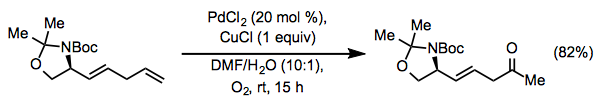
In Wacker oxidations, Lewis-basic functionality in the allylic or homoallylic position relative to the reactive alkene can induce unpredictably high site selectivity. Unprotected allylic alcohols form mixtures of methyl ketone and aldehyde products under standard Tsuji-Wacker conditions. To overcome this issue, the catalyst Pd(Quinox)Cl2 was created; in the presence of this catalyst, allylic alcohols react preferentially to yield methyl ketones. In this process, the oxygen source is tert-Butyl hydroperoxide (TBHP).
(10)
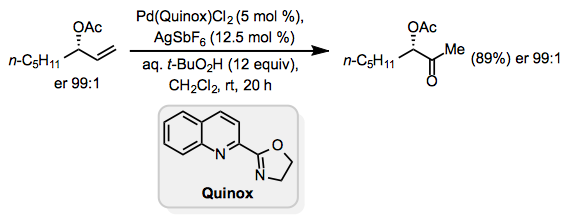
Protected homoallylic alcohols generate the methyl ketone preferentially, but the findings for unprotected homoallylic alcohols are more variable.
Protected allylic amines produce larger quantities of aldehyde than equivalent protected allylic alcohols; these findings show that aldehyde generation is caused by palladium coordination to the Lewis base. The Wacker oxidation can be made totally selective for aldehydes by using a phthalimide group in the allylic position
(11)
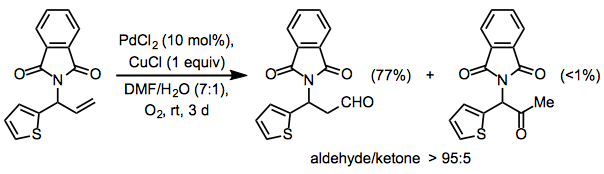
Unless the substrate contains an allylic or homoallylic Lewis base, site selectivity in internal alkene reactions is minimal. Electron-withdrawing groups can guide oxidation to the alkene's position, but they require relatively high catalyst loadings.
(12)
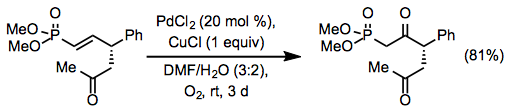
Wacker cyclization can occur in the presence of an adequately positioned nucleophile in the substrate, yielding an allylic or vinylic ether. Protected amino nucleophiles like sulfonamides participate in the aza-Wacker cyclization.
(13)
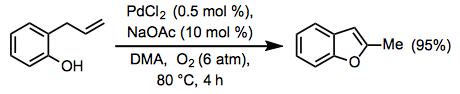
Acetal production can occur when an alcohol is utilised in stoichiometric or solvent proportions, or when a diol is present in the substrate. Acetal production, like ketone creation, is a Markovnikov selective process. In the presence of (R,R)-2,4-pentanediol, a Michael acceptor produces an acetal at the site.
(14)
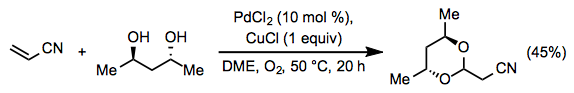
Synthetic Applications
In organic synthesis, the Wacker oxidation has been widely utilised. The installation of terminal alkenes can be accomplished in a variety of ways, and the methyl ketone products of the process can be easily evolved into more complicated compounds. Furthermore, the reaction is aerobic and straightforward to do.
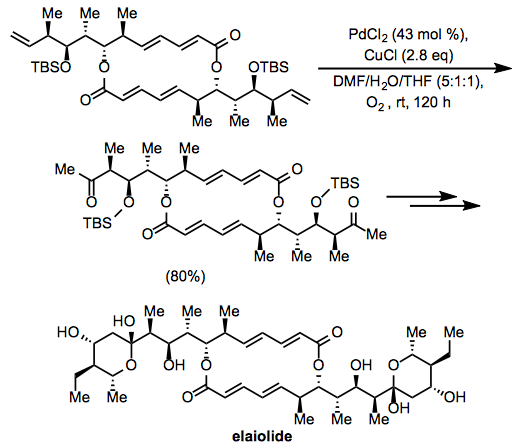
During the synthesis of the macrolide elaiolide, the Wacker oxidation was used. In a C2-symmetric intermediate, oxidation at two terminal alkenes yielded a diketone, which was then elaborated to the symmetric natural product. The internal alkenes and esters in the intermediate, for example, appeared to have no effect on the oxidation.
(15)
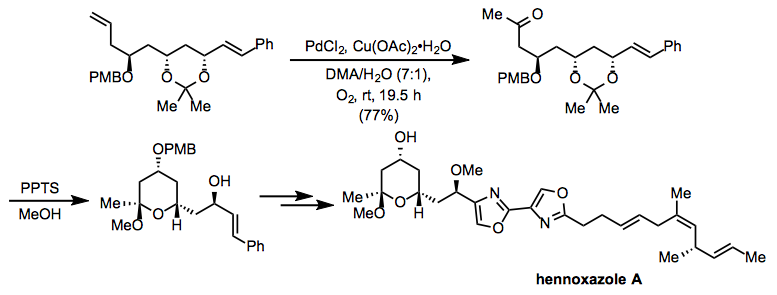
During the synthesis of hennoxazole A, a variation of Tsuji-Wacker conditions using copper(II) acetate was used. Deprotection under acidic circumstances after oxidation resulted in the intramolecular production of an acetal present in the target molecule.
(16)
Comparison to Other Methods
There are few true Wacker oxidation alternatives (that is, other hydration-oxidation processes). Oxymercuration followed by palladium trans metalation is one relatively severe approach.
(17)
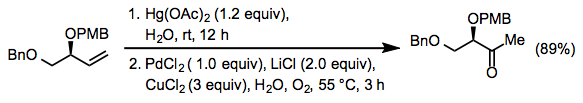
Experimental Conditions and Procedure
Typical Conditions
Tsuji-Wacker conditions are an excellent starting point for this reaction's experimental circumstances. An organic co-solvent (typically DMF) is added to water, together with 10 mol% PdCl2, stoichiometric CuCl, and 1 atmosphere of oxygen gas. Copper(I) salts are preferable to copper(II) salts because the former reduce chloride concentration in solution, promoting syn-hydroxypalladation. Copper(I) is rapidly oxidised to copper(II) by dissolved oxygen; to guarantee that this oxidation is complete, a 30-minute interval is utilised prior to the introduction of the substrate. When Tsuji-Wacker conditions fail, a variety of reaction condition adjustments can be used.
Wacker oxidations can be carried out without the use of a co-oxidant by using high oxygen pressure and a careful selection of solvent. Direct O2-coupled oxidations are also promoted by some ligands.
Wacker oxidations can also use peroxides as the oxygen source; these reactions do not require a co-oxidant. Silver hexafluoroantimonate acts as a chloride scavenger in the Pd(Quinox)/TBHP system.
(18)
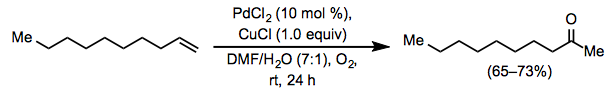
A magnetic stirring bar and a pressure-equalizing dropping funnel were connected to a 100-mL, 3-necked, round-bottomed flask holding 1-decene (4.2 g, 30 mmol, 1.0 equiv). PdCl2 (0.53 g, 3 mmol, 0.1 equiv), CuCl (2.97 g, 30 mmol, 1.0 equiv), and aqueous DMF (DMF/H 2O = 7:1, 24 mL) were added to the flask. An oxygen-filled balloon was placed over one neck, and the mixture was agitated at rt to facilitate oxygen uptake, with the other exits firmly stoppered and wired down. After 1 hour, the 1-decene (4.2 g, 30 mmol) was added over a 10-minute period, and the solution was vigorously agitated at room temperature under an oxygen balloon. Within 15 minutes, the solution's colour changed from green to black, then gradually reverted to green. After 24 hours, the mixture was put into 100 mL of cool 3 N HCl and extracted with five 50 mL ether fractions. The extracts were mixed together and washed with 50 mL of saturated aqueous NaHCO3 solution, 50 mL of brine, then dried over anhydrous magnesium sulphate in that order. Evaporation was used to remove the solvent, and the residue was distilled using a 15 cm Vigreux column to yield 2-decanone as a colourless oil (3.0–3.4 g, 65–73%): IR (neat) 1722 cm–1, bp 43.5 °C (1 mm Hg); 1H NMR (CCl4) 2.37 (t, J = 7 Hz, 2H), 2.02 (s, 3H), 0.7–1.8 (m, 15H).
Qualitative Cation Analysis
Qualitative analysis, a process for determining the identity of metal ions present in a mixture, can be used to determine the composition of relatively complicated mixes of metal ions (rather than quantitative information about their amounts). The method for isolating and identifying more than 20 common metal cations from a single solution involves selectively precipitating only a few types of metal ions at a time under specific circumstances. As seen in Figure, successive precipitation processes become increasingly less selective until practically all of the metal ions are precipitated.
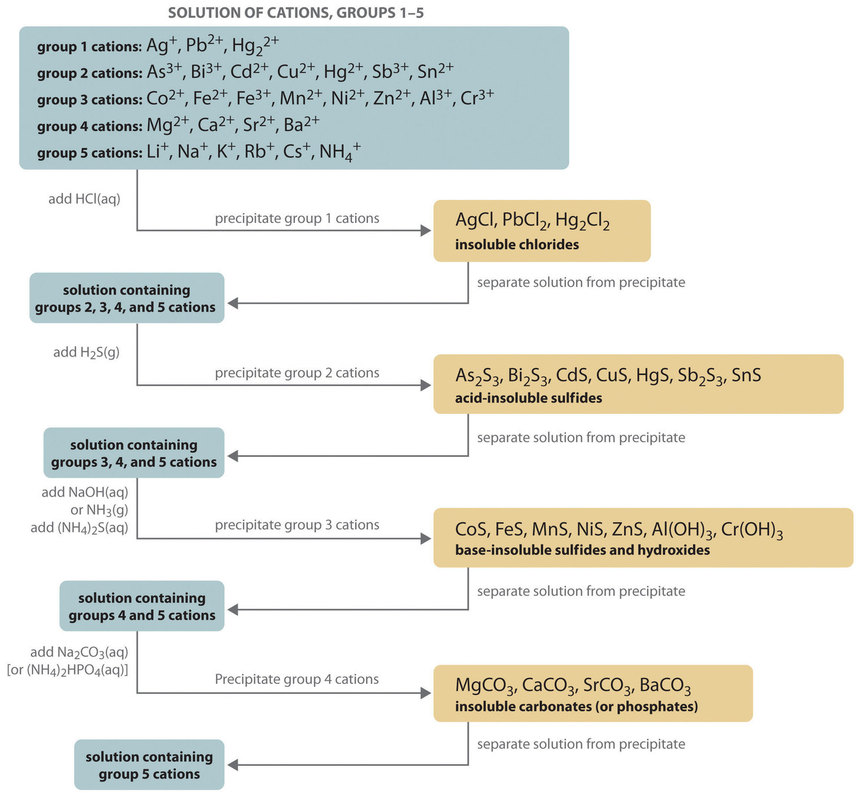 Figure: Steps in a Typical Qualitative Analysis Scheme for a Solution That Contains Several Metal Ions
Figure: Steps in a Typical Qualitative Analysis Scheme for a Solution That Contains Several Metal Ions
Group 1: Insoluble Chlorides
Only Ag+, Pb2+, and Hg22+ create chlorides that precipitate from water; the rest are soluble in water. In a qualitative analysis, the first step is to add roughly 6 M HCl, which causes AgCl, PbCl2, and/or Hg2Cl2 to precipitate. These cations are not present in large levels if no precipitate forms. Filtration or centrifugation might be used to collect the precipitate.
Group 2: Acid-Insoluble Sulfides
Next, the acidic solution is saturated with H2S gas. Only those metal ions that form very insoluble sulfides, such as As3+, Bi3+, Cd2+, Cu2+, Hg2+, Sb3+, and Sn2+, precipitate as their sulfide salts under these acidic conditions. All others, such as Fe2+ and Zn2+, remain in solution. Once again, the precipitates are collected by filtration or centrifugation.
Group 3: Base-Insoluble Sulfides (and Hydroxides)
Ammonia or NaOH is now added to the solution until it is basic, and then (NH4)2S is added. This treatment removes any remaining cations that form insoluble hydroxides or sulfides. The divalent metal ions Co2+, Fe2+, Mn2+, Ni2+, and Zn2+ precipitate as their sulfides, and the trivalent metal ions Al3+ and Cr3+ precipitate as their hydroxides: Al(OH)3 and Cr(OH)3. If the mixture contains Fe3+, sulfide reduces the cation to Fe2+, which precipitates as FeS.
Group 4: Insoluble Carbonates or Phosphates
The metal ions that form insoluble carbonates and phosphates are the next to be removed from solution. Insoluble carbonates precipitate and are collected when Na2CO3 is added to the basic solution that remains after the precipitated metal ions have been removed. Alternatively, the same metal ions precipitate as insoluble phosphates when (NH4)2HPO4 is added.
Group 5: Alkali Metals
At this point, all metal ions that produce water-insoluble chlorides, sulphides, carbonates, or phosphates have been eliminated. Alkali metals (Li+, Na+, K+, Rb+, and Cs+) and ammonium (NH4+) are the only common ions that may survive. To neutralise the ammonium ion and produce NH3, we take a second sample from the original solution and add a small amount of NaOH. (Because we added ammonium to that sample in earlier phases, we can't utilise the same sample we used for the first four groups.) The odour of ammonia produced or a litmus paper test can both be used to detect it. To identify sodium, a flame test on another original sample is employed, which creates a brilliant yellow colour. In flame tests, the other alkali metal ions give off distinct colours, allowing them to be distinguished if only one is present.
Additional strategies, such as creating complex ions, changing the pH of the solution, or increasing the temperature to redissolve part of the solids, are used to separate metal ions that precipitate together. For instance, the precipitated metal chlorides of group 1 cations, such as Ag+, Pb2+, and Hg22+, are all water insoluble. However, because PbCl2 is much more soluble in hot water than the other two chloride salts, adding water to the precipitate and boiling the resulting slurry will dissolve any PbCl2 that may be present. If Pb2+ was present in the original sample, isolating the solution and adding a little amount of Na2CrO4 solution to it will result in a brilliant yellow PbCrO4 precipitate.

Figure: When a small amount of Na2CrO4 solution is added to a sample containing Pb2+ ions in water, a bright yellow precipitate of PbCrO4 forms. From Wikipedia.
As another example, treating the precipitates from group 1 cations with aqueous ammonia will dissolve any AgCl because Ag+ forms a stable complex with ammonia: [Ag(NH3)2]+. In addition, Hg2Cl2 disproportionates in ammonia

To form a black solid that is a mixture of finely divided metallic mercury and an insoluble mercury(II) compound, which is separated from solution:
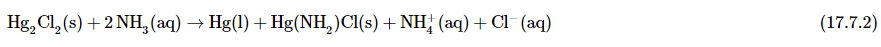
Any silver ions in the solution are then detected by adding HCl, which reverses the reaction and produces a white AgCl precipitate that darkens slowly when exposed to light:
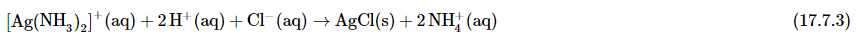
Separating and identifying the various components of the other groups uses similar but significantly more sophisticated reactions.
Solubility Product Principle and Qualitative Analysis
In qualitative analysis, the solubility product principle is used to determine the composition of a compound by separating ions in a solution.
Qualitative Analysis
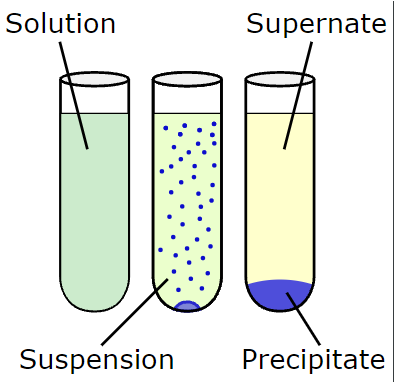
Chemical precipitation:
The formation of a solid in a solution or inside another solid during a chemical reaction or by diffusion in a solid is known as precipitation. In qualitative chemical analysis, precipitation is used.
The elemental composition of inorganic compounds is determined using traditional qualitative inorganic analysis, a method of analytical chemistry. Its primary purpose is to detect ions in an aqueous solution. The solution is treated with a variety of reagents to see if it reacts like certain ions do, which can result in colour changes, solid formation, and other visible changes.
• Solubility Product Principle and Qualitative Analysis
Solubility-product constants can be used to devise methods for separating ions in a solution by selective precipitation. Selective precipitation is used to form a solid with one of the ions in solution without disturbing the other ions. You can continue this method to effectively separate all of the ions in a solution. The entire traditional qualitative-analysis scheme is based on the use of these equilibrium constants to determine the correct precipitating ions and the correct strategy.
• Qualitative Analysis of Cations
Six groups of cations are commonly used. Each group has a common reagent for separating itself from the solution. Because cationic analysis is based on the ions' solubility products, only if separation is done in a specific order can meaningful results be obtained. Because some ions from one group may react with the reagent from another group, this is the case. Both Ba2+ and Sr2+, for example, will solidify when reacting with the SO42- ion. As a result, before using the SO42- ion for selective precipitation in a solution containing both Ba2+ and Sr2+, it is necessary to perform mathematical calculations.
Key Takeaway:
* In qualitative analysis, a solution is treated with various reagents to test for the presence of certain ions.
*Solubility – product constants can be utilised to develop strategies for precipitating ions selectively in a solution.
Cations are usually separated into six groups, each with its own reagent for separating them from the solution.
Systematic Analysis of Anions
Aim:
Using a variety of tests, determine the presence of anionic radicals in an inorganic mixture of salts.
Theory:
In qualitative analysis, acidic and basic radicals present in inorganic salts are detected and identified. Acids and bases, or acidic oxides with a base or basic oxides, react to produce inorganic salts.
The reaction of acids and bases, or acidic oxides with a base or basic oxides, can be seen in the following examples.
NaOH + HCl → NaCl + H2O
CO2 + 2NaOH → Na2CO3 + H2O
KOH + HNO3 → KNO3 + H2O
2NaOH + H2SO4 → Na2SO4 + 2H2
Because they have defined geometrical shapes, the majority of organic compounds are crystalline solids. They are usually made up of radicals, which are negatively charged particles or ions.
In the salt analysis, two fundamental principles are of great use
• Solubility Product
• Common Ion Effect
1. Solubility Product:
In an equation representing electrolyte dissociation at a given temperature when the solution is saturated, the solubility product is defined as a product of ion concentrations raised to a power equal to the number of occurrences of ions. Only if the solution is saturated is the solubility product the ionic product.
2. Common Ion Effect:
The common ion effect is a phenomenon that occurs when a small amount of strong electrolyte containing a common ion is added to any weak electrolyte, suppressing the degree of dissociation. The ionisation of the weak electrolyte acetic acid is suppressed by adding a strong electrolyte sodium acetate containing the common acetate ion.
Materials Required:
• Test tubes
• Boiling tubes
• Test tube holder
• Test tube stand
• Corks
• Filter paper
• Delivery tube
• Reagents
Apparatus Setup:
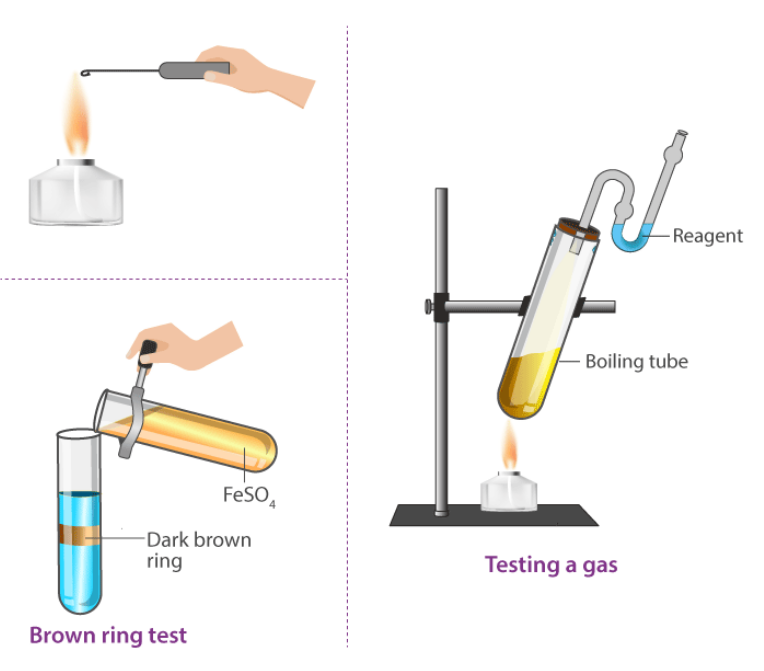
Systematic Analysis of Anions
Procedure:
S.No | Experiment | Observation | Inference |
1 | Preliminary reactions | Given salt is colourless | Fe2+, Fe3+, Ni2+ and Co2+ ions are absent. |
Appearance | If the given salt is green | May be Fe2+, Ni2+, Cu2+ | |
If the given salt is brown | May be Fe2+ | ||
If the given salt is pink | May be Co2+. Mn2+ | ||
If the given salt is blue | Cu2+ | ||
2 | Action of Heat: Take a small quantity of the salt in a dry test tube, heat it gently. | A colorless gas with a pungent odor that turns moist red litmus blue paper. | May be NH4+ salt. |
Reddish brown vapors are obtained that turn acidified ferrous sulfate brown paper. | May be NO3– | ||
In cold, the substance is white and in hot, yellow. | May be (Zn)2+ | ||
3 | Flame Test: Add a drop of Con.HCl to a small amount of salt in a watch glass and put it in a paste. Put the paste into the base of the non-luminous bunsen burner with the help of a glass rod | (a) Apple green colour flame. (b) Crimson red colour flame. (c) Brick red colour flame. (d) Bluish green flame | May be Ba2+ May be Sr2+ May be Ca2+ May be Cu2+ |
Identification of Anions from Volatile Products:
S.No | Experiment | Observation | Inference |
4 | Action of dilute.H2SO4: Add 1 or 2ccs of dilute H2SO4 to a small portion of the salt in a test tube and warm it gently. | Brisk effervescence is obtained from colorless, odorless gas that turns lime water. | Anion is Carbonate CO32- |
Colorless gas with an odor of rotten eggs is obtained, turning lead acetate black paper. | Sulphide anion is present. | ||
Colorless gas with the odor of burning sulfur that turns acidified dichromate green. | Sulfate anion is present. | ||
Reddish brown gas is obtained with a fishy odor that turns acidified brown ferrous sulphate. | Nitrate anion is present. | ||
Colorless vinegar-flavored gas is obtained. | May be acetate anion. | ||
No characteristic observation. | Absence of above mentioned anions. | ||
5 | Action of Con. H2SO4: Add 2-3 ccs of Con. H2SO4 to a small amount of salt taken in a test tube and heat it gently. | Reddish-brown vapors that turn moist red paper fluorescent. | May be bromide anion. |
Colorless gas with a pungent smell that provides dense white fumes with a dipped glass rod in NH4OH solution. | May be chloride anion. | ||
Violet colored vapors that turn blue or violet starch paper. | May be iodide anion. | ||
Reddish-brown vapors turning brown ferrous sulfate paper into acidified. | May be nitrate anion. | ||
No characteristic observation. | Absence of all above-mentioned anions. | ||
6 | Action of Con. H2SO4 with Cu turnings: Mix with a few Cu bits a small amount of the salt taken in a test tube, add 2 – 3 ccs of H2SO4 and heat it. | It is observed that copy evolution of reddish brown gas turns acidified ferrous sulfate paper brown. | Nitrate anion is present |
No reddish brownvapours. | Absence of nitrate anion. | ||
7 | Action of Con. H2SO4 with MnO2: Add an equal amount of MnO2 to a small amount of salt in a test tube and add a few ccs of Con. H2SO4 and heat gently. | A greenish yellow gas turns violet (or) blue starch iodide paper. | May be chloride anion. |
Reddish brown vapors are obtained that turn moist fluorescent red paper. | May be bromide anion. | ||
Violet vapors are obtained that turn starch paper blue (or) violet. | May be iodide anion. | ||
No characteristic colouredvapours are obtained. | Absence of all above mentioned anions. | ||
8 | Action of NaOH: Add a few ccs of 10% NaOH solution to a pinch of salt taken in a test tube and warm it gently. | It is obtained a colorless gas with a pungent smell that produces dense white fumes with a glass rod dipped in HCl. | May be ammonium ion. |
There is no characteristic gas released from ammonium. | Absence of ammonium. |
Sodium Carbonate Extract:
Confirmatory Tests for CO32-, S2–, SO32-, NO2– and CH3COO– anions:
Confirmatory anion testing is carried out using water extract when salt is water-soluble and using sodium carbonate extract when salt is water-insoluble. Confirmation of CO32– is done using aqueous salt solution or using solid salt as such, as carbonate ions are contained in the sodium carbonate extract. Extract of water is produced by dissolving salt in water.
Preparation of Sodium Carbonate Extract:
In a boiling tube or porcelain dish, place one gramme of salt. 15 mL distilled water + 3 g solid sodium carbonate Cook for 10 minutes after removing the contents. Cool, strain, and label the filtrate as a sodium carbonate extract in a test tube.
S.No | Experiment | Observation | Inference |
9 | Silver nitrate test: Add dilute HNO3 to a portion of the sodium carbonate extract until the effervescence stops. Add a few drops of AgNO3 solution in excess, 2 – 3nos. | Curdy white NH4OH soluble precipitate. | May be chloride anion. |
Pale yellow precipitate in NH4OH that is sparingly soluble. | May be bromide anion. | ||
Insoluble yellow precipitation in NH4OH. | May be iodide anion. | ||
No characteristics precipitate. | Absence of all above mentioned anions. | ||
10 | Barium Chloride test: Add BaCl2 solution to about one or two ccs of the extract (after acetic acid neutralization and CO2 boiling). Add dilute hydrochloric acid to a portion of the ppt above. | An insoluble white precipitate in HCl. | The anion is SO42- |
A HCl soluble white precipitate. | The anion is SO32- | ||
No characteristics precipitate. | Absence of SO42- and SO32-. | ||
11 | Lead acetate test: Add lead acetate solution to about one or two ccs of the extract (after acidification with acetic acid, CO2 boiling off and cooling). | White ppt, soluble in excess of the solution of ammonium acetate. | Presence of SO42- is confirmed. |
12 | Ferrous Sulphate Test: (Also called Brown ring test) Add dilute H2SO4 in drops to about 1 or 2cc of extract until the effervescence stops. And a few drops in excess add two to three drops of freshly prepared FeSO4 solution. Keep in a slanting position the test tube, add Con. H2SO4 without interfering with the solution | At the liquid junction, a brown ring is obtained. | Nitrate anion (NO3–) is present. |
No brown ring formed | Nitrate anion (NO3–) is absent. | ||
13 | Ferric chloride test: Take the extract in a test tube for about 1 or 2ccs and add a neutral FeCl3 solution. If necessary, filter and split the solution or filtrate into two parts: | Deep red colouring produced. | Acetate anion is confirmed. (CH3COO–) |
1. Add dil. HCl | Red colouring is gone. | CH3COO– is confirmed. | |
2. Add water and boil to the second part | Reddish brown precipitate. | CH3COO– is confirmed. | |
14 | Calcium Chloride Test: In a test tube, add dil to a portion of the extract of sodium carbonate. Boil off CO2 with acetic acid. Add a few drops of a solution of calcium chloride. | A white calcium oxalate precipitate is obtained. | Confirms the presence of oxalate anion. |
Incorporate dil. HNO3 to the hot and white ppt | Precipitate dissolves. |
| |
15 | Ethyl Acetate Test: Add a few drops of ethanol to a pinch of salt taken in a test tube, followed by 1 or 2ccs of H2SO4. Heat it gently and cool it down. Version Na2CO3. | There is a pleasant fruity odor. | It is confirmed that anion acetate is present. |
Observations and Inference:
S.No | Anions | Observation |
1 | CO32‒ (Carbonate anion) | CO2 gas is evolved with brisk effervescence with dilute sulphuric acid, which turns lime water milky. |
2 | S2‒ (Sulphide anion) | Add a drop of solution for sodium nitroprusside. Appears purple or violet colouring. |
3 | SO32‒ (Sulfite anion) | A white precipitate is formed with a barium chloride solution that dissolves in dilute hydrochloric acid and sulfur dioxide gas also develops. |
4 | SO42‒ (Sulfate anion) | Take 1 mL of salt water extract in water or sodium carbonate and add BaCl 2 solution after acidification with dilute hydrochloric acid. Insoluble white precipitate in conc. HCl or HCl. It gets HNO3. |
5 | NO2‒ (Nitrite anion) | Add a few drops of iodide potassium solution and a few drops of starch solution, acidify with acetic acid. Blue colour appears. |
6 | NO3‒ (Nitrate anion) | Take in a test tube 1 mL of salt solution in water. Add a conc of 2 mL. Mix thoroughly with H2SO4. Cool the mixture under the tap. Add freshly prepared ferrous sulfate without shaking on the sides of the test tube. At the junction of the two solutions, a dark brown ring is formed. |
7 | Cl‒ (Chloride anion) | In a test tube, take 0.1 g of salt, add a pinch of manganese dioxide and 3-4 drops of conc. Acid with sulfuric acid. Heat the mixture of reactions. Greenish yellow chlorine gas that is detected by its strong odor and bleaching action |
8 | Br‒ (Bromide anion) | Take 0.1 g of salt in a test tube with a pinch of MnO2. Add conc.sulphuric acid and heat with 3-4 drops. There is an evolution of intense brown fumes. |
9 | I‒ (Iodide anion) | Take in a test tube 1 mL of salt solution in water. Add a conc of 2 mL. Mix thoroughly with H2SO4. Cool under the tap the mixture. Add freshly prepared ferrous sulfate without shaking on the sides of the test tube. At the junction of the two solutions, a dark brown ring is formed. |
10 | PO43‒ (Phosphate anion) | Acidify the extract of sodium carbonate or salt solution in water and con HNO3 and add solution of ammonium molybdate and heat to boil. A precipitate canary yellow is formed. |
11 | C2O42‒ (Oxalate anion) | Take 1 mL of acetate acidified water extract or sodium carbonate extract and add a solution of calcium chloride. An insoluble white precipitate is formed in the solution of ammonium oxalate and oxalic acid but soluble in dilute hydrochloric acid and diluted nitric acid. |
12 | CH3COO‒ (Acetate anion) | Add 1 mL and 0.2 mL conc of ethanol. Heat and H2SO4. The presence of acetate ion is confirmed by fruity odor. |
Results:
The given salt contains ________ (CO32‒, S2‒, SO32‒, SO42‒, NO2‒, NO3‒, Cl‒, Br‒, I‒, PO43‒, C2O42‒, CH3COO‒) anion.
Precautions:
1. Before using any reagents or chemicals, read the label on the bottle carefully. If a reagent isn't labelled, never use it.
2. When working in a chemical lab, always wear an apron, eye protection, and hand gloves.
3. Always be cautious when sniffing chemicals or vapours. Gently fan the vapours towards your nose at all times.
4. Mixing chemicals and reagents isn't always necessary. Chemicals are not discernible.
5. Pour the acid into the water for dilution. Water should never be acidified.
6. Never put sodium metal down the drain or into the trash.
7. Due to the fact that the test tube is heated, proceed with caution. When heating or adding reagents, never point the test tube at yourself or your neighbours.
8. Always dilute acid with water. Adding acid to water is never a good idea.
9. Maintain a clean work environment. Papers and glass should never be thrown down the drain. Always use a dustbin for this.
10.Explosive substances, flammable substances, toxic gases, electrical appliances, glass products, flames, and hot substances should all be avoided.
11. Use the smallest amount of reagents possible. Excessive reagent use not only wastes chemicals but also harms the environment.
12. After you've finished your laboratory work, always wash your hands.
Qualitative Analysis of Cations
The Theory
What is Qualitative Analysis?
Qualitative analysis is a method of Analytical chemistry that deals with the determination of elemental composition of inorganic salts. It is mainly concerned with the detection of ions in an aqueous solution of the salt.
The common procedure for testing any unknown sample is to make its solution and test this solution with various reagents for the ions present in it. Testing with various reagents gives characteristic reaction of certain ions, which may be a colour change, a solid formation or any other visible changes. There are separate procedures for detecting cations and anions, called the Cation Analysis and Anion Analysis.
Let us discuss about the Qualitative Analysis of Cations.
Let us discuss about the Qualitative Analysis of Cations.
Qualitative Analysis of Cations
Preliminary Tests
Some preliminary tests needs to be done before doing the analysis of cations.
(a) Physical Appearance: Color and Smell
The colour, smell, and density of the unknown salt are all examined physically. The test isn't very accurate, but it does help identify some coloured cations. Some ions, such as ammonium, have a distinct smell that aids in their identification.
(b) Charcoal Cavity Test
This test is based on the fact that metallic carbonates decompose to produce corresponding oxides when heated in a charcoal cavity. In the cavity, the oxides show up as a coloured incrustation or residue. In some cases, the formed oxides are partially reduced to metal, resulting in metallic beads or scales.
Examples:
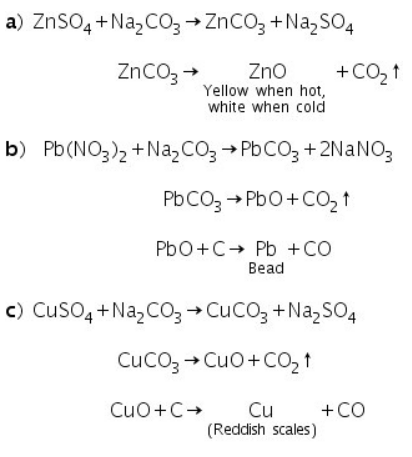
(c) Cobalt Nitrate Test (Ash test)
The salts that leave white residue in the charcoal cavity test are subjected to this test. This test is based on the fact that metallic carbonates decompose to produce corresponding oxides when heated in a charcoal cavity.
The test is based on the fact that heating causes cobalt nitrate to decompose into cobalt oxide, CoO. This reacts with the metallic oxides in the charcoal cavity, which are present as a white residue, to produce coloured compounds.
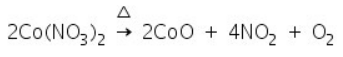
A while residue of MgO is left behind when a magnesium salt undergoes a charcoal cavity test, for example. This forms a double salt with the formula MgO.CoO, which is pink in colour, after being treated with cobalt nitrate and then heated.
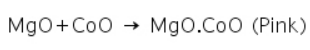
Other examples are:
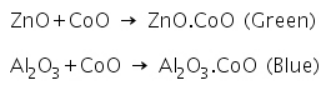
In addition to metallic oxides, phosphates and borates also react with cobalt oxide to form Co3(PO4)2 and Co3(BO3)2 that are blue in colour.
(d) Flame Test
Certain salts form chloride when they react with concentrated HCl, which is volatile in a non-luminous flame. The flame takes on the colour of their vapours. The presence of certain cations can be reliably determined by this colour. To carry out this test, a platinum wire is used to introduce the paste of the mixture with conc.HCl into the flame.
(e) Borax Bead Test
On heating, borax, Na2B4O7.10H2O, fuses and loses crystallisation water. It expands into a fluffy white porous mass, which then melts into a colourless liquid, resulting in a clear transparent glassy bead made of boric anhydride and sodium metaborate.
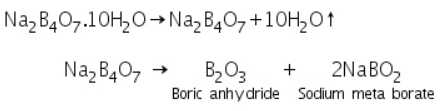
Boric anhydride is not a flammable substance. It forms a distinctive coloured bead of metal metaborate when it reacts with a coloured metallic salt. Metaborates in various oxidation states of metals are formed in those cases where different coloured beads are obtained in oxidising and reducing flames. Copper forms blue copper metaborate in an oxidising flame, for instance.
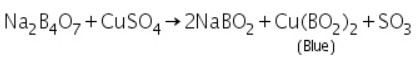
In reducing flame, cupric metaborate is reduced to metallic copper, which is red and opaque.

Identification of Cations (Basic Radicals)
Analysis of Group Zero
This group includes ammonium ion (NH4+).
Chemical reactions involved in Group Zero analysis
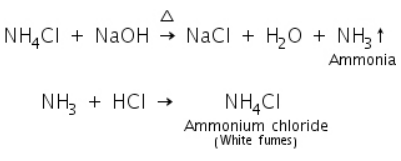
(a) Sodium hydroxide test
Ammonia gas is formed when ammonium salt is heated with conc. NaOH, resulting in white fumes with dil.HCl due to the formation of NH4Cl.
(b) Nessler's reagent test
Tha ammonia gas formed by the reaction of ammonium ions with NaOH reacts with Nessler's reagent to form a brown precipitate of H2N.HgO.HgI.
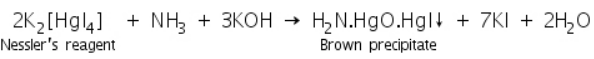
Analysis of Group I
Pb2+, Ag+, and Hg22+ are all members of this group. Only Pb2+ will be discussed in this section. This group's reagent is diluted HCl.
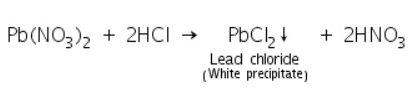
Chemical reactions involved in Group I analysis
Pb2+ will precipitate as lead chloride when HCl is added to the solution, which is soluble in hot water. The precipitate settles as PbCl2, which is less soluble in cold water, as it cools.
Confirmatory tests of Lead (II) ion (Pb2+)
(a) Potassium iodide test
In hot water, lead chloride (formed by reacting lead salts with dilute HCl) reacts with potassium chromate solution to form a yellow precipitate of lead chromate.
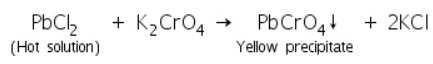
Analysis of Group II
This group includes Pb2+ and Cu2+ in IIA Group and As3+ in IIB Group.
Chemical reactions involved in Group II Analysis:
Passing of H2S gas through the acidified original solution will precipitate the radicals Pb2+,Cu2+,As3+ as their sulphides.
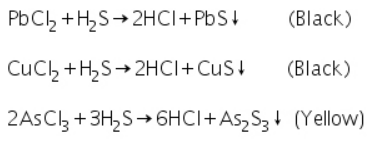
Confirmatory tests for Lead (II) ion (Pb2+)
Due to the formation of soluble lead nitrate, the black PbS precipitate formed in group analysis dissolves in 50 percent nitric acid. Lead sulphate precipitates after sulphuric acid is added to soluble lead nitrate.
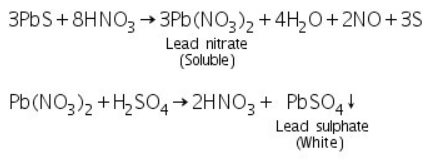
(a) Potassium iodide test
The yellow precipitate of lead iodide is formed when soluble lead nitrate (formed by reacting PbS with 50 percent Conc. HNO3) reacts with potassium iodide.

Confirmatory tests for Copper (II) ion (Cu2+)
CuS precipitate formed in group analysis dissolves in 50% nitric acid, yielding a blue solution when excess NH4OH is added.
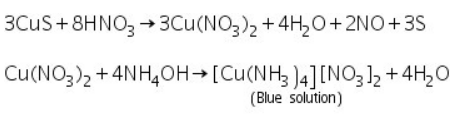
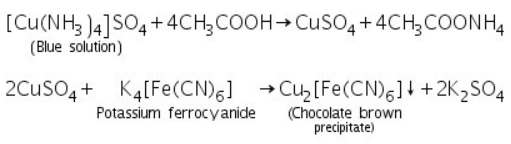
(a) Potassium ferrocyanide test
With potassium ferrocyanide solurion, the blue solution above yields a chocolate brown precipitate.
(b) Potassium iodide test
The white precipitate in this test is caused by the formation of cuprous iodide, while the brown colour of the solution is caused by the liberation of iodine.
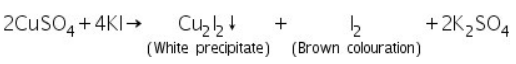
Confirmatory tests of Arsenic (III) ion (As3+)
The yelllow residue of As2S3 formed in the group analysis is dissolved in conc.HNO3 forming arsenic acid.
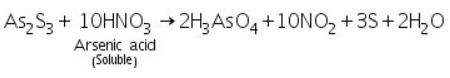
(a) Ammonium molybdate test
Arsenic acid (formed by dissolving As2S3 in conc.HNO3) reacts with ammonium molybdate to produce ammonium arseno molybdate, a yellow precipitate.

(b) Magnesia mixture test
Mg(NH4)2AsO4 is formed when arsenic acid (formed by dissolving As2S3 in conc.HNO3) reacts with magnesia mixture to form a white precipitate.
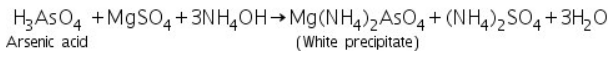
Analysis of Group - III
The cations present in this group are Fe2+, Fe3+, Cr3+ and Al3+. We will look at only Fe2+/ Fe3+ and Al3+.
Chemical Reactions Involved Group III Analysis
By adding ammonium hydroxide in the presence of ammonium chloride, the cations in this group precipitate as hydroxides. NH4OH in the presence of NH4Cl is thus the group reagent for this group.
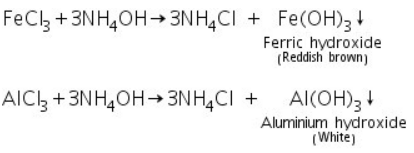
Confirmatory tests of Ferric ion (Fe3+)
Due to the formation of soluble ferric chloride, the reddish brown precipitate of ferric hydroxide is dissolved in HCl.
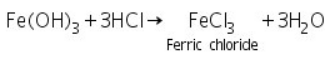
(a) Potassium ferrocyanide test
Prussian blue Ferric ferrocyanide is formed when ferric chloride (made by dissolving ferric hydroxide in HCl) reacts with potassium ferrocyanide.
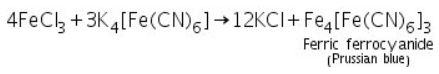
(b) Potassium sulphocyanide test
Ferric chloride (which is made by dissolving ferric hydroxide in HCl) reacts with potassium sulphocyanide to produce ferric sulphocyanide, which is blood red in colour.
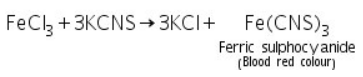
Confirmatory tests of Aluminium (III) ion ( Al3+)
(a) Lake test
In dil. HCl, the aluminium hydroxide generated in the group analysis dissolves to form soluble aluminium chloride. Aluminum hydroxide is generated when aluminium chloride interacts with ammonium hydroxide. This precipitate has taken on the blue colour of litmus solution.
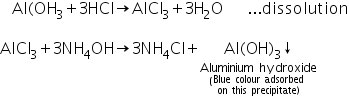
(b) Charcoal cavity/Cobalt nitrate test
Aluminium oxide produced in the charcoal cavity test combines with CoO in the cobalt nitrate test to form Al2O3.CoO, resulting in a blue mass.
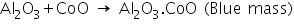
Analysis of Group - IV
CO2+, Ni2+, Mn2+, and Zn2+ are the radicals found in this group. By passing H2S gas through the salt's ammonical solution, these are precipitated as sulphide. H2S gas in the presence of NH4Cl and NH4OH is the group reagent for this group.
In Group IV Analysis, a chemical reaction is involved.
When H2S gas is passed through a group III solution, the radicals Co2+, Ni2+, Mn2+, and Zn2+ precipitate as sulphides. The presence of black ppt. (CoS or NiS) suggests the presence of cobalt or nickel. Manganese is represented by buff-colored ppt (MnS), whereas zinc is represented by filthy white ppt (ZnS).

Confirmatory tests of Cobalt (II) ion (Co2+)
(a) Potassium nitrite test
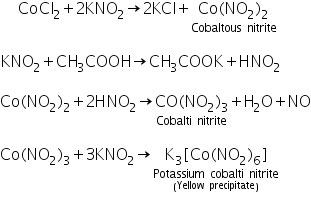
In the presence of acetic acid, the cobalt ion interacts with potassium nitrite to generate a yellow precipitate of potassium cobalti nitrite.
(b) Ammonium thiocyanate ether test
The synthesis of ammonium cobalti thiocyanate results in a blue colour in the ethereal layer when ether and an ammonium thiocyanate crystal are combined.
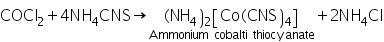
(c) Borax bead test
Co2+ ion creates blue beads in both oxidising and reducing flames in this experiment.
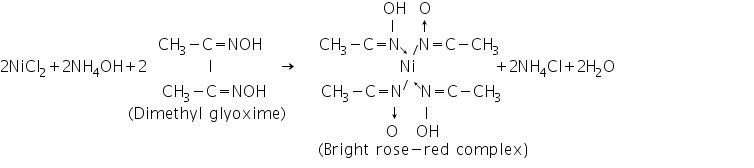
Nickel (II) ion (Ni2+) confirmatory assays (a) Dimethyl glyoxime test
Nickel2+ ions react with dimethyl glyoxime to generate Ni(dmgH)2, a vivid rose-red Nickel-Dimethyl glyoxime complex.
(b) Sodium hydroxide-bromine water test
Nickelic hydroxide is formed when Ni2+ ions react with an excess of NaOH and bromine water to generate a black precipitate.
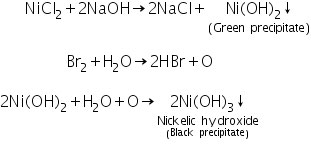
(c) Borax bead test
In oxidising and reducing flames, Ni2+ ions generate brown bead and grey bead, respectively.
Manganese (II) ion (Mn2+) confirmatory tests Manganese sulphide generated in the group analysis dissolves in dil.
Manganese chloride is formed by HCl, and H2S is removed by boiling.
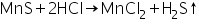
(a) Sodium hydroxide - bromine water test
Manganese chloride (produced by dissolving MnS in HCl) interacts with excess NaOH to form manganese hydroxide, a white precipitate.
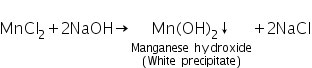
Due to oxidation to brown manganic hydroxide MnO(OH)2, the white precipitate of manganese hydroxide turns brown when bromine water is added.
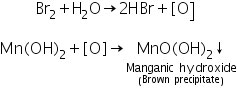
(b) Lead peroxide test
Due to the creation of HMnO4, manganese sulphide generated in the group analysis reacts with Conc. HNO3 and lead peroxide to form a pink-colored solution.
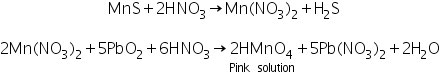
(c) Borax bead test
In oxidising flames, Mn2+ ions form pinkish beads, while in reducing flames, they produce colourless beads.
Zinc (II) ion (Zn2+) confirmatory tests
In dil, the white ZnS precipitate generated during the group analysis dissolves. HCl reacts with zinc chloride to generate zinc chloride, and H2S is released during the boiling process.
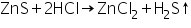
(a) Sodium hydroxide test
Zinc chloride (made by dissolving ZnS in dilute HCl) interacts with sodium hydroxide to produce zinc hydroxide, which is a white precipitate.

(b) Potassium ferrocyanide test
Zinc chloride (made by dissolving ZnS in dilute HCl) interacts with potassium ferrocyanide to generate zinc ferrocyanide, which is white or bluish white in colour. 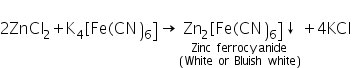
(c) Charcoal cavity/Cobalt nitrate test
Zinc oxide produced in the charcoal cavity test combines with CoO in the cobalt nitrate test to make ZnO.CoO, resulting in a greenish residue.
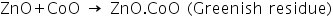
Analysis of Group - V
Ba2+, Sr2+, and Ca2+ are the three radicals that make up Group V. The carbonates of these cations are precipitated. This group's group reagent is (NH4)2CO3 in the presence of NH4Cl and NH4OH.
In Group V Analysis, a chemical reaction is involved.
The carbonates of Ba2+, Sr2+, and Ca2+ precipitate when (NH4)2CO3 is introduced to a salt solution containing NH4Cl and NH4OH.
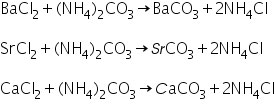
Barium (II) ion (Ba2+) confirmation
Due to the generation of soluble barium acetate, the white precipitate of barium carbonate generated in the group analysis dissolves in hot dil. Acetic acid.
Barium (II) ion (Ba2+) confirmation
Due to the generation of soluble barium acetate, the white precipitate of barium carbonate generated in the group analysis dissolves in hot dil. Acetic acid.
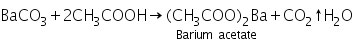
(a) Potassium chromate test
The yellow precipitate of barium chromate is created when barium acetate (made by dissolving barium carbonate in dilute acetic acid) combines with potassium chromate.
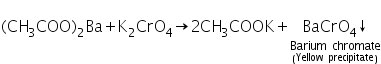
(b) Flame test
The flame takes on a grassy green hue when barium is present.
Strontium (II) ion (Sr2+) confirmation
Due to the generation of soluble strontium acetate, the white precipitate of strontium carbonate generated in the group analysis dissolves in hot dil. Acetic acid.
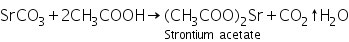
(a) Ammonium sulphate test
Strontium acetate (produced by dissolving strontium carbonate in dilute acetic acid) interacts with ammonium sulphate to produce strontium sulphate, a white precipitate.
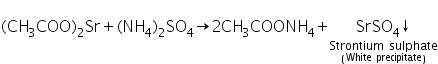
(b) Flame test
The flame is ruby red due to the presence of strontium.
Calcium (II) ion (Ca2+) confirmation
Due to the development of soluble calcium acetate, the white precipitate of calcium carbonate generated in the group analysis dissolves in hot dil. Acetic acid.
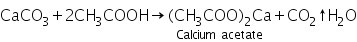
(a) Ammonium oxalate test
Calcium acetate (produced by dissolving calcium carbonate in dilute acetic acid) interacts with ammonium oxalate to produce calcium oxalate, a white precipitate.
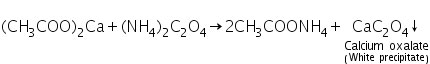
(b) Flame test
Calcium imparts brick red colour to the flame.
Analysis of Group - VI
Chemical Reaction involved in Confirmation of Mg2+
(a) Ammonium Phosphate test
Mg2+ ions react with ammonium phosphate in presence of NH4Cl and NH4OH to form white precipitate of magnesium ammonium phosphate.

(b) Charcoal cavity/Cobalt nitrate test
In this test, magnesium oxide produced in the charcoal cavity test reacts with CoO in cobalt nitrate test to produces a pink mass due to the formation of MgO.CoO.
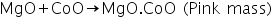
What is the Common Ion Effect?
In a solution containing numerous species associating with each other via a chemical equilibrium process, raising the concentration of one of the ions dissociated in the solution by introducing another species carrying the same ion will result in an increase in the degree of ion association.
The common ion effect is seen when gaseous hydrogen chloride is fed through a sodium chloride solution, causing the NaCl to precipitate due to the excess chloride ions in the solution (brought on by the dissociation of HCl).
Transition metal compounds do not exhibit this effect. The d-block elements have a proclivity for forming complex ions, which explains why. Cuprous chloride, an insoluble in water compound, demonstrates this. The CuCl2– complex ion is soluble in water and can be dissolved in water by adding chloride ions.
Effect on Solubility
• In this subsection, we'll look at how the addition of a common ion affects the solubility of a salt in a solution.
• The common ion effect can be used to obtain drinking water from chalk or limestone-containing aquifers (underground layers of water mixed with permeable rocks or other unconsolidated materials). The hardness of the water is reduced by adding sodium carbonate (chemical formula Na2CO3) to it.
• The common ion effect is used in water treatment to precipitate calcium carbonate (which is sparingly soluble) from the water by adding sodium carbonate (which is highly soluble).
• This addition of sodium carbonate produces a finely divided calcium carbonate precipitate with a very pure composition. As a result, the CaCO3 precipitate is a valuable by-product that can be used in the production of toothpaste.
• Because soaps are sodium salts of carboxylic acids with long aliphatic chains (fatty acids), the common ion effect can be seen in the soap-making process. In order to reduce the soap's solubility, sodium chloride is added to the solution.
• However, water with a sufficient amount of Na+ ions, such as seawater and brackish water, can inhibit the action of soaps by lowering their solubility and thus their effectiveness.
PH and the Common-Ion Effect
The pH of a buffer solution changes when the conjugate ion of a buffer solution (solution containing a base and its conjugate acid, or acid and its conjugate base) is added to it due to the common ion effect.
• When acetic acid and sodium acetate are both dissolved in the same solution, acetate ions are produced. Sodium acetate, on the other hand, completely dissociates, whereas acetic acid only partially ionises. Because acetic acid is a weak acid, sodium acetate is a strong electrolyte, this is the case.
• According to Le Chatelier's theory, sodium acetate's new acetate ions aid in the suppression of acetic acid ionisation, shifting the equilibrium to the left. The pH of the solution is raised because the dissociation of acetic acid is reduced.
• As a result, when compared to an acetic acid solution, the common ion solution containing acetic acid and sodium acetate has a higher pH and is thus less acidic.
Thus, this article discusses the common ion effect, its impact on the solubility of a salt in a solution, and its impact on the pH of a solution.
Key takeaway:
• common ion: An ion that is in common to both salts in a solution.
• common ion effect: A decrease in the solubility of an ionic compound as a result of the addition of a common ion.
Solubility Product Principle and Qualitative Analysis
Qualitative Analysis
The elemental composition of inorganic compounds is determined using classical qualitative inorganic analysis, a method of analytical chemistry. Its primary purpose is to detect ions in aqueous solutions. The solution is treated with a variety of reagents to see if certain ions cause colour changes, solid formation, or other visible changes.
Solubility Product Principle and Qualitative Analysis
Methods for separating ions in a solution by selective precipitation can be devised using solubility-product constants. Selective precipitation is a technique for forming a solid out of one of the ions in solution while leaving the other ions alone. You can use this method to separate all of the ions in a solution effectively. The use of these equilibrium constants to determine the correct precipitating ions and strategy is the foundation of the entire traditional qualitative-analysis scheme.
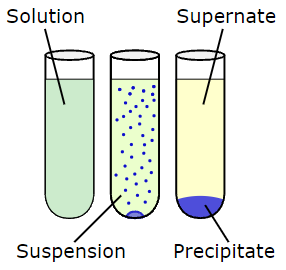
Chemical precipitation
Precipitation is the formation of a solid in a solution or within another solid as a result of a chemical reaction or diffusion within a solid. Qualitative chemical analysis employs precipitation.
Qualitative Analysis of Cations
Six groups of cations are commonly used. Each group has a common reagent for separating itself from the solution. Because cationic analysis is based on the ions' solubility products, only if separation is done in a specific order can meaningful results be obtained. Because some ions from one group may react with the reagent from another group, this is the case. Both Ba2+ and Sr2+, for example, will solidify when reacting with the SO42- ion. As a result, prior to selecting the SO42- ion for selective precipitation in a solution containing both Ba2+ and Sr2+, it is necessary to perform mathematical calculations.
Example:
The concentrations of barium chloride (BaCl2) and strontium chloride (SrCl2) in a solution are 0.010 M and 0.020 M, respectively (SrCl2). Is it possible to use concentrated sodium sulphate (Na2SO4) solution to selectively precipitate either Ba2+ or Sr2+? Which ion is going to be the first to precipitate? (Assume the Na2SO4 solution is so concentrated that the volume change in the Ba-Sr solution can be ignored for the sake of simplicity.)
Solution:
The barium sulfate solubility is given by:
Ksp = [Ba2+][SO42-] = 1.5 x 10-9
With 0.010 [latex]\frac{moles}{liter} [/latex]of Ba2+, precipitation of barium sulfate will not occur until the sulfate ion concentration increases to:
[SO42-] = 0.010 = 1.5 x 10-7
Strontium sulfate will precipitate when the sulfate concentration is:
This signifies that as the sulfate ion (SO42-) is added to solution and its concentration increases, barium will precipitate first.
What is interfering radicals in salt analysis?
What are the radicals interfering with the process? What role do they play in the systematic separation of cationic radicals? Why should they be deleted before the analysis of the third grade? Why don't they participate in the first or second group's analysis? Anionic radicals such as oxalate, tartrate, fluoride, borate, and phosphate obstruct DNA synthesis. They form a combination with ammonium chloride and ammonium hydroxide, which are also reagents in the IIIrd gr group. This causes incomplete precipitation of IIIrd group cations in an alkaline media, as well as early precipitation of IVth and Vth group cations. Let's take a look at it and see what we can learn about it. Metals including oxalate, tartrate, fluoride, borate, silicate, and phosphate are soluble in acidic environments.
Remember that the medium for the first and second analyses is acidic (diluted HCl), so they don't interfere. However, the third group's reagents, ammonium chloride and ammonium sulphide, cause the medium to become alkaline. Interfering radicals interfere with the solubility product of cations, causing their precipitation to occur prematurelyor incompletely.
In an acidic environment, these salts produce oxalic acid, phosphoric acid, hydrofluoric acid, boric acid, and tartaric acid. Barium oxalate, for example, reacts with HCl to produce oxalic acid.
BaC2O4 + 2HCl ⟶ BaCl2 + H2C2O4
Because these interfering acids are weak, they do not completely dissociate and remain in solution in their unionised form. Between dissociated and non-dissociated acids, equilibrium develops.
H2C2O4 ⇌ 2H+ + C2O42-
Hydrochloric acid is a strong acid and is ionised completely.
HCl ⟶ H+ + Cl-
Hydrogen ions act as a common ion among them, and a higher concentration of H+ suppresses interfering acid ionisation. As a result, the ionic product of C2O42- and Ba2+ does not exceed the solubility product of barium oxalate, and Ba2+ is retained in solution as barium oxalate. As long as the medium is acidic enough, interfering radicals do not interfere. When we add the 3rd group reagent ammonium hydroxide NH4OH to make the medium alkaline, however, OH- ions combine with H+ and neutralise them. This lowers the concentration of H+ ions, which advances the dissociation equilibrium of the interfering acid and raises the concentration of C2O42-. As a result, the ionic product of C2O42- and Ba2+ exceeds the solubility product of barium oxalate, and Ba2+ precipitates in the third group, which is actually part of the fourth.
The solution may contain one or more interfering radicals. They must be removed in the following order: oxalate and tartrate first, then borate and fluoride, silicate next, and finally phosphate.
Key takeaway:
• Add concentrated HCl and again evaporate to dryness. On heating with HCl fluoride forms hydrofluoric acid and Borate forms orthoboric acid which evaporate on heating. Extract the residue with dilute HCl and filter. Use this filtrate for analysis of 3rd group or use for removal of other interfering radicals.
• Interfering radicals are oxalate, tartrate, fluoride, borate and phosphate all of them are anionic radicals. They form a complex with third group reagents which are ammonium chloride and ammonium hydroxide.
References:
1. Das Asim K., Fundamentals of Inorganic Chemistry, Vol. II, CBS Publications, 2nd Ed. 2010.
2. Selected Topic in Inorganic Chemistry, Mallick, Madan and Tuli, S. Chand Publisher. 17th Ed. 2010.
3. Mehrotra R.C. And Singh, A. Organometallic Chemistry, New Age International Publishers, 2nd Edn, 2000.
4. Gupta B. D. And Elias A. J., Basic Organometallic Chemistry, 2nd Edn., University Press (2013).




