Unit - 2
Enzymes
You might find some enzyme-containing household goods if you browse around the laundry room cupboard. At first glance, this may appear unusual; enzymes are found in the body and have no business being in a stain remover or drain cleaning. Enzymes, on the other hand, are just molecules that excel at doing specialised activities. Enzymes are quite good at breaking certain bonds and replacing them with new ones in these domestic applications. They're breaking down larger molecules in the process, which could result in a stain or a delayed drain. Those larger molecules break down into smaller molecules that are easier to wash away.
Enzymes are what kind of molecules they are. They're unique proteins. Proteins play a variety of roles in life, including providing structure and catalysing processes. Proteins that catalyse processes are known as enzymes. Biopolymers are a type of molecule that includes proteins. Large molecules made up of smaller molecules that have been bound together are known as biopolymers. Amino acids are the tiny building components of proteins. The fact that amino acids are chiral, or have extremely unique three-dimensional shapes, such as a left-handed glove or a right-handed glove, is one of their most fundamental characteristics.
• Proteins are made up of chiral amino acids
• Enzymes are proteins
• Enzymes are large, chiral molecules that are superb at what they do.
What is the actual size of these proteins? Titin is the most abundant protein, and it is responsible for some of muscle's elasticity. Titin is made up of almost 35,000 different amino acids. Because an amino acid's molecular weight is around 100 Da (100 Daltons or 100 amu), titin has a molecular weight of around 35 million Da.
Ticin, on the other hand, is a structural protein. Let's take a closer look at an enzyme. Trypsin is a very common digestive enzyme that is found in the stomach. The image below shows the results of a structural determination experiment with pig trypsin. It has a molecular weight of roughly 45,000 Da and is made up of roughly 450 amino acids.

That image depicts every atom in trypsin except the hydrogens, which are too tiny to be seen. Even so, it's quite crowded. Enzymes and other proteins are frequently depicted in simplified, cartoon form. The beta sheets and alpha helices, which are secondary structures inside trypsin, are highlighted in the cartoon model below. You can get a better feel of the shape of things in the cartoon model. Gaps and grooves may be seen in the protein, as well as the chirality of the helices.
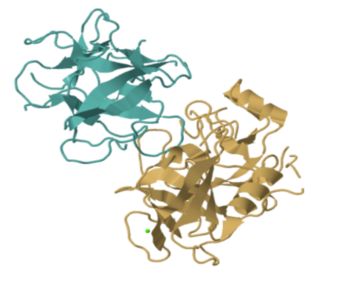
Trypsin belongs to the hydrolase family of enzymes. Hydrolases are enzymes that use a water molecule to break bonds. Trypsin (yep, it's a cannibal) breaks particular amide or peptide bonds in other proteins, transforming the carbonyl side of the bond into a carboxylic acid. This begins the process of breaking down proteins in food, which is an important aspect of digestion.
However, how does trypsin accomplish this?
At the most fundamental level, enzymes bond to their substrates, perform chemical reactions on them, and then release them. Binding the substrate is a crucial step, and it's one of the reasons why enzyme chirality matters. Some enzymes have a very narrow range of substrates to which they bind. They may, for example, bind one enantiomer but not the other.
Trypsin is also exceedingly specific, as it only breaks the amide link between arginine and lysine residues. The reaction it catalyses isn't always specific to that particular peptide bond, but trypsin binds to that location specifically.
The "lock and key" mechanism is a term used to describe the role of specific binding in enzymes. When the correct key is inserted, enzymes are unlocked or turned on, causing the enzyme to leap into activity. We'll look at how enzymes bind to their substrates and carry out their reactions in more detail in the sections that follow.
2.2.1 Enzymes Classification:
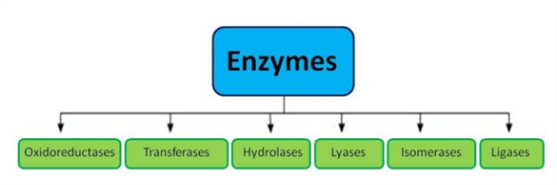
Previously, enzymes were given names based on who discovered them. The classification got more comprehensive as more research was done.
Enzymes are split into six functional classes, according to the International Union of Biochemists (I U B), and are categorised depending on the type of reaction they catalyse. Hydrolases, oxidoreductases, lyases, transferases, ligases, and isomerases are the six types of enzymes.
Listed below is the classification of enzymes discussed in detail:
Types | Biochemical Characteristic |
Oxidoreductases | The oxidation reaction in which electrons tend to go from one form of a molecule to the other is catalysed by the enzyme Oxidoreductase. |
Transferases | Transferase’s enzymes aid in the translocation of functional groups between acceptors and donors. |
Hydrolases | Hydrolases are hydrolytic enzymes that catalyse the hydrolysis reaction by cleaving and hydrolyzing the bond by adding water. |
Lyases | Water, carbon dioxide, or ammonia is added across double bonds to form double bonds, or it is removed to form double bonds. |
Isomerases | Isomerase enzymes catalyse structural alterations in a molecule, resulting in a change in the shape of the molecule. |
Ligases | Ligase’s enzymes are known to be involved in the catalysis of ligation reactions. |
- Oxireductase
By withdrawing or adding electrons from or to their substrates, these enzymes catalyse oxidation or reduction. Dehydrogenase, oxidase, and reductase are examples of enzymes.
- Transferase
These enzymes move a group of molecules from one substrate to another. Transaminase, for example.
- Hydrolases
These enzymes break certain covalent bonds and hydrolyze water to break bigger molecules into smaller ones. Carbohydrase, for example.
AB + HOH -> AH+BOH (AB Substrate)
- Lyases
Without hydrolysis, these enzymes catalyse the breaking of particular covalent bonds and the elimination of groups. Histidine decarboxylase, for example.
- Isomerases
Isomerases are enzymes that catalyse the rearranging of atoms inside a molecule to generate isomers. Phosphohexoisomerase, for example.
- Ligases
Ligases catalyze the formation of C - C, C - S, C - O and C - N bonds. The energy for the reaction is derived by the hydrolysis of ATP. e.g., pyruvate carboxylase.
2.2.2 Properties of enzymes
1. The enzymes, like inorganic catalysts, are active in extremely little concentrations and stay unaltered when the reaction is completed.
2. Enzymes have a fairly narrow range of action. i.e., a specific enzyme catalyses a specific sort of reaction by acting on a specific substrate. However, the same process may be performed by many enzymes at times. Isoenzymes are enzymes that work in this way.
3. Enzymes are extremely heat sensitive. The temperature at which an enzyme is most active is referred to as its optimal temperature. Both above and below the optimal temperature, enzyme activity decreases. The catalytic component of the enzyme is made up of proteins. When the temperature rises beyond 50°C, the protein denatures, and the enzyme loses its action.
4. The enzyme's catalytic property is affected by its pH. The optimal pH is the pH at which the enzyme activity is highest. The optimal pH value for various enzymes is variable. The pH of most intracellular enzymes will be around neutral. Tripsins are active in an alkaline environment, diastase in a neutral environment, and pepsin in an acidic environment.
5. Inhibitors have a strong effect on enzymes. An inhibitor is a chemical compound that binds to enzymes and prevents them from catalysing reactions. Cyanides, for example.
A-><-B.
The reactions catalysed by enzymes are usually reversible, depending on the needs of the cell.
• Although a few catalytically active RNA molecules have been discovered, nearly all enzymes are proteins.
• When contrasted to chemical reactions, enzyme catalysed reactions normally take place under comparatively mild conditions (temperatures considerably below 100oC, air pressure, and neutral pH).
• Enzymes are catalysts that speed up the rate of a chemical reaction without changing the reaction itself.
• Enzymes are quite particular in terms of the substrates they operate on and the products they produce.
• Enzyme activity can be controlled, fluctuating in reaction to substrate or other molecule concentrations.
• They work in the body under stringent temperature and pH parameters.
2.3.1 Enzymes and activation energy
A catalyst is a material that speeds up a chemical reaction without becoming a reactant. Enzymes are catalysts for biological reactions that occur in living organisms. Although most enzymes are proteins, certain ribonucleic acid (RNA) molecules also function as enzymes.
Enzymes play a crucial role in lowering the activation energy of a process—that is, the amount of energy required to start the reaction. Enzymes work by binding to reactant molecules and holding them in such a way that the chemical bond-breaking and bond-forming processes take place more readily.
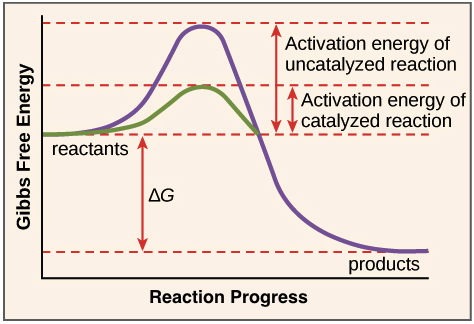
The path of a reaction with and without a catalyst is depicted in this reaction coordinate diagram. The activation energy is lower with the catalyst than without it. The catalyst, on the other hand, has no effect on the reaction's G.
To clarify one point, enzymes do not modify the G value of a process. That is, they have no effect on whether a reaction is overall energy-releasing or energy-absorbing. This is due to the fact that enzymes have no effect on the free energy of the reactants or products.
Enzymes, on the other hand, lower the energy of the transition state, which products must pass through in order to become reactants. In the diagram above, the transition state is at the top of the energy "hill."
2.3.2 Active sites and substrate specificity
An enzyme will grip (bind) to one or more reactant molecules to catalyse a process. The enzyme's substrates are these compounds.
One substrate can be broken down into numerous products in some reactions. In others, two substrates are combined to form a bigger molecule or portions are swapped. In fact, there is almost certainly an enzyme to speed up any biological reaction you can think of!
The active site (where the catalytic "activity" takes place) is the region of the enzyme where the substrate binds.
A substrate enters the enzyme's active site. The enzyme-substrate complex is formed as a result of this. Following that, the reaction takes place, converting the substrate to products and establishing an enzyme products complex. The products then exit the enzyme's active site.

Proteins are made up of units called amino acids, and the active site of enzymes, which are proteins, obtains its features from the amino acids that make it up. These amino acids can have large or small side chains, acidic or basic, hydrophilic or hydrophobic side chains.
The active site's amino acid composition, as well as their relative placements in 3D space, give it a distinct size, shape, and chemical behaviour. An enzyme's active site is specifically designed to bind to a specific target—the enzyme's substrate or substrates—and assist them in undergoing a chemical reaction thanks to these amino acids.
2.3.3 Environmental effects on enzyme function
Active sites are extremely sensitive to changes in the enzyme's environment because they are finely tailored to aid a chemical reaction. The active site and enzyme function may be affected by the following factors:
- The temperature of the room. Higher temperatures, whether enzyme-catalyzed or not, result in faster reaction rates. Increased or decreased temperature outside of a safe range, on the other hand, can disrupt chemical bonds in the active site, rendering them less well-suited to bind substrates. Very high temperatures (for animal enzymes, above 40 degrees C, 104 degrees F) may cause an enzyme to denature, losing its shape and activity.
- The ph level. Enzyme function can also be affected by pH. The acidic or basic characteristics of active site amino acid residues are critical for catalysis. PH changes can alter these residues, making it difficult for substrates to bind. Enzymes perform well within a specified pH range, and excessive pH values (acidic or basic) can cause enzymes to denature, just like temperature.
2.3.4 Induced fit
The interaction between an enzyme's active site and its substrate isn't as simple as two jigsaw pieces fitting together (as scientists formerly believed, in the "lock-and-key" paradigm).
Instead, when an enzyme attaches to its substrate, it alters shape somewhat, resulting in a tighter match. Induced fit refers to the enzyme's adjustment to fit the substrate tightly.
The induced fit model of enzyme catalysis is depicted. When a substrate binds to the active site, it changes shape slightly, tightening its grip on the substrate and prepares to catalyse the reaction. The products are released from the active site and diffuse away once the reaction has occurred.
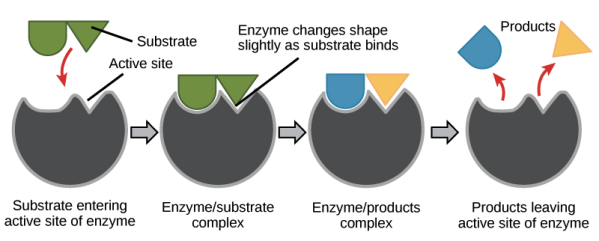
We know that when an enzyme connects to its substrate, it decreases the reaction's activation energy, allowing it to proceed more quickly. But you might be wondering, what exactly does the enzyme do to the substrate to lower the activation energy?
The enzyme determines the response. By bringing two substrates together in the correct direction, some enzymes can speed up chemical reactions. Others provide a reaction-friendly environment inside the active site (for example, one that's slightly acidic or non-polar). The enzyme-substrate complex can also help to attain the transition state by bending substrate molecules in a way that aids bond-breaking and lowers activation energy.
Finally, by participating in the chemical reaction, some enzymes reduce activation energy. As part of the reaction process, active site residues may create transient covalent connections with substrate molecules.
The word "temporary" is crucial here. At the end of the reaction, the enzyme will always return to its natural state—it will not remain linked to the reacting molecules. In fact, one of enzymes' distinguishing characteristics is that they are unaffected by the reactions they catalyse. When an enzyme finish catalysing a process, it simply releases the product (or products) and prepares for the next catalysis cycle.
2.4.1 Mechanism of Trypsin
Because the active sites of trypsin and chymotrypsin are identical, the mechanisms of both are identical. This process consists of four steps:
Step 1
To begin, the hydrogen on serine's OH is removed, resulting in a negative charge on the oxygen. The extra electrons can target the peptide bond, causing the negative charge to assault it:
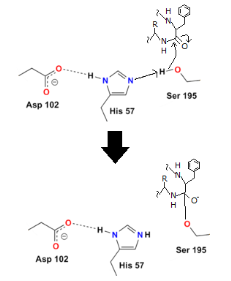
Step 2
The oxygen's electrons can easily repair a carbon-oxygen double bond, but then another carbon bond must be formed. In this situation, the peptide's carbon-nitrogen link is broken. The histidine nitrogen provides a hydrogen to the nitrogen. The first protein residue from the enzyme has now been released:
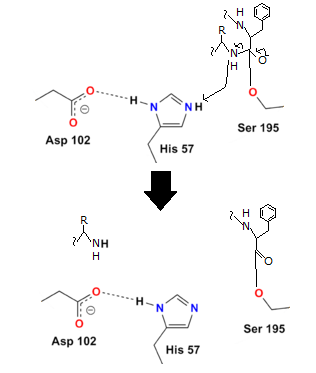
Step 3
Although the peptide bond has been broken, the reaction has not been completed since the other half of the protein (the part containing the basic amino acid) remains linked to the enzyme. The final two steps assist the enzyme in releasing the second half of the protein.
Water is added in order to do this. The hydrogen from the water binds to the histidine, while the OH binds to the carboxyl carbon, preventing oxygen from taking on a negative charge:
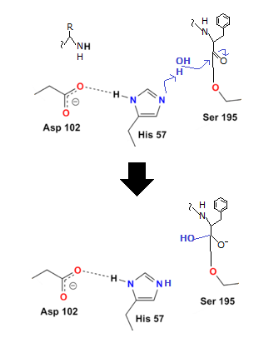
2.5.1 Factors Affecting Enzyme Activity:
• Biochemical reactions occur in all living species' bodies and are required for growth, tissue repair, and energy production. These events are referred to as "metabolism," and they occur often in living organisms. • All reactions that occur in living organisms require substantial activation energy to take place, and if they stop working, the organism will die. A catalyst ensures that chemical reactions occur quickly and that energy activation is reduced, reducing the cell's energy consumption. The enzymes act as a catalyst.
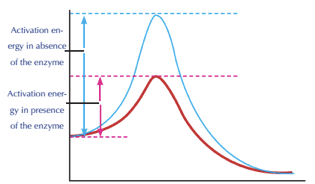
Enzyme activation energy
• Enzymes are big protein molecules that act as biological catalysts. They hasten chemical reactions within the cell. The enzyme is made up of a series of amino acids that link together to form a polypeptide chain.
• Enzymes are chemical catalysts similar to other catalysts. They take part in the response but are unaffected. In other words, they increase the rate of chemical reactions within cells without consuming them. The hydrogen ion concentration (pH) and temperature have an impact on enzymes. In comparison to other catalysts, enzymes are very specific, with each enzyme specialised for a single reactant chemical. This reactive substance is referred to as substrate, and it is designed to perform only one or a few reactions. Enzymes reduce the amount of energy required to initiate a reaction. These are the enzyme's most important features taken together.
• Several factors influence the speed of an enzyme's action, including the enzyme's concentration, the substrate's concentration, temperature, hydrogen ion concentration (pH), and the presence of inhibitors.
Factor 1: Concentration of Enzyme
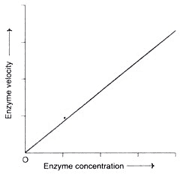
Concentration of enzymes
• As the enzyme concentration rises, the reaction velocity rises in lockstep. This feature is utilised in the diagnosis of diseases to determine the activity of serum enzymes.
Factor 2: Concentration of Substrate
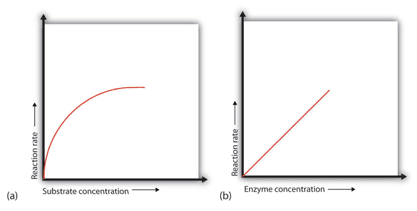
Concentration of Substrate Enzyme
• When a certain amount of enzyme is present, the rate of enzymatic reaction increases as the substrate concentration increases until a limiting rate is achieved, after which additional increases in the substrate concentration have no effect on the reaction rate. Because there is so much substrate at this phase, almost all of the enzyme active sites have substrate bound to them. In other words, the substrate has saturated the enzyme molecules. The extra substrate molecules are unable to react until the substrate that has already been bound to the enzymes has reacted and been released (or been released without reacting).
Factor 3: Effect of Temperature
• Because enzymes are proteins, they are particularly sensitive to temperature variations. When compared to regular chemical reactions, enzyme activity occurs across a small temperature range. As you can see, each enzyme is more active at a particular temperature.
• As the temperature rises over the ideal temperature, the enzyme activity gradually decreases until it reaches a temperature at which the enzyme activity totally ceases due to a change in its natural composition. If the temperature falls below the ideal temperature, the enzyme activity decreases until the enzyme reaches a minimum temperature where it has the least activity. At 0°C, enzyme activity ceases altogether, but when the temperature increases again, the enzyme is reactivated.
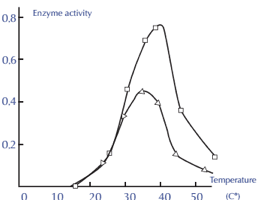
Enzyme activity and temperature
Factor 4: Effect of pH
• The best way to determine the concentration of hydrogen ion (H+) in a solution is to use the potential of hydrogen (pH). It can also tell if a liquid is acidic, basic, or neutral. Acids are liquids with a pH less than 7, whereas bases or alkalines are liquids with a pH more than 7. At 25 degrees Celsius, pH 7 liquids are neutral and have the same acidity as pure water. The pH indicators can be used to determine the pH of any solution.

PH Indicators
• Proteins with acidic carboxylic groups (COOH–) and basic amino groups are known as enzymes (NH2). As a result, changing the pH value has an impact on the enzymes. The ideal pH for an enzyme is the pH at which it functions most efficiently. The enzyme activity diminishes until it stops operating if the pH is lower or higher than the optimum pH. Pepsin, for example, functions at a low pH, indicating that it is highly acidic, whereas trypsin acts at a high pH, indicating that it is basic. The majority of enzymes operate at a pH of 7.4.
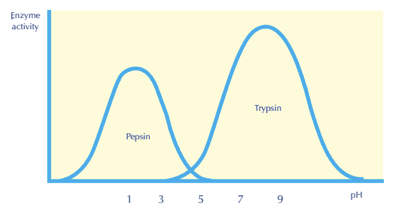
Enzyme PH activity
Factor 5: Effect of Activators
Some of the enzymes require certain inorganic metallic cations, like Mg2+, Mn2+, Zn2+, Ca2+, Co2+, Cu2+, Na+, K+ etc., for their optimum activity. Rarely, anions are also needed for enzyme activity, e.g., a chloride ion (CI–) for amylase.
2.6.1 Coenzymes & Cofactors:
Coenzymes and cofactors are substances that aid in the proper functioning of an enzyme or protein. Coenzymes are organic compounds that attach loosely to an enzyme's active site to enhance substrate recruitment, whereas cofactors do not attach to the enzyme. Cofactors are "assistant molecules" that might be inorganic or organic. Metal ions, for example, are frequently required to improve the rate of catalysis of a specific enzyme-catalyzed reaction. These coenzymes and cofactors contribute in the catalysis of a variety of cellular metabolic reactions, serving both structural and functional roles.
Bio Vision has a great portfolio of assays in this category which include assays for NAD, NADP, FAD, Acetyl CoA, ATP and many such molecules. The assays are simple, rapid, and compatible with a large number of sample types.
Enzymes require extra components to work properly. Cofactors and coenzymes are two types of molecules that fall within this category. Metallic ions are cofactors, while non-protein organic compounds are coenzymes. Both sorts of assistance molecules can be tightly coupled to the enzyme or can only be attached when the substrate binds.
Cofactors are found in about 30% of mature proteins. They are commonly folded into an enzyme and are involved in the catalytic action of the enzyme. Magnesium is required for more than 300 enzymes in the human body, including DNA polymerase. The magnesium ion assists in the creation of the phosphodiester bond on the DNA backbone in this scenario. Other typical cofactors include iron, copper, cobalt, and manganese.
Coenzymes are nonprotein, organic aid molecules for enzymes, and many vitamins are coenzymes. Biotin, a B vitamin, is required for a number of enzymes that transport carbon dioxide from one molecule to another. Biotin, vitamin A, and other vitamins must be obtained from our diet because human cells are incapable of producing them.
2.6.2 Functions of Coenzymes
Apoenzymes are enzymes that don't have a cofactor. Enzymes cannot catalyse processes adequately without coenzymes or cofactors. In fact, it's possible that the enzyme won't work at all. An organism's ability to sustain life will be hampered if reactions cannot occur at the regular catalytic rate.
An enzyme becomes a holoenzyme, or active enzyme, when it acquires a cofactor. Active enzymes convert substrates into the products that an organism need to perform key chemical and physiological processes. Coenzymes, like enzymes, can be reused and recycled multiple times without affecting the rate or efficacy of the process. They bind to a part of an enzyme's active site, allowing the catalytic reaction to proceed. The coenzyme can no longer connect to the active site when an enzyme is denatured by excessive temperature or pH.
2.6.3 Examples of Coenzymes
Most organisms are unable to synthesise sufficient levels of coenzymes spontaneously. They are instead introduced to an organism in one of two ways:
- Vitamins
Many, but not all, coenzymes are vitamins or are generated from vitamins. If a person's vitamin consumption is insufficient, he or she will lack the coenzymes required to catalyse processes. Coenzymes are produced by water-soluble vitamins, which include all B complex vitamins and vitamin C. Nicotinamide adenine dinucleotide (NAD) and coenzyme A are two of the most important and widely used vitamin-derived coenzymes.
When converted into its two alternate forms, NAD, which is produced from vitamin B3, serves as one of the most significant coenzymes in a cell. When NAD loses an electron, it forms NAD+, a low-energy coenzyme. When NAD gets an electron, it becomes the high-energy coenzyme NADH.
NAD+ is primarily responsible for transferring electrons required for redox processes, particularly those involved in the citric acid cycle (TAC). Other coenzymes, like as ATP, are produced by TAC. Mitochondria become less functioning and supply less energy for cell operations when an organism has a NAD+ shortage.
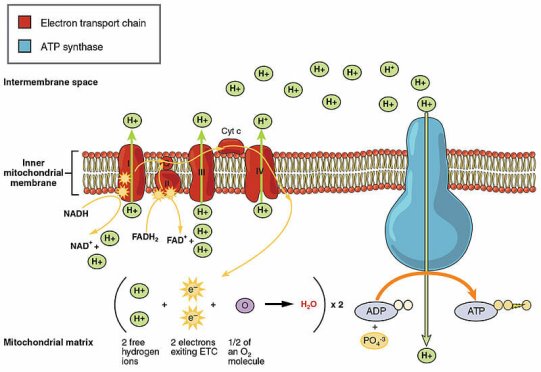
NADH is generated when NAD+ obtains electrons during a redox process. NADH, often known as coenzyme 1, serves a variety of purposes. In fact, it is the most important coenzyme in the human body because it is required for so many functions. This coenzyme is principally responsible for transporting electrons during reactions and producing energy from meals. The electron transport chain, for example, can only start with the transfer of electrons from NADH. NADH deficiency causes energy deficiencies in cells, resulting in weariness. This coenzyme is also known as the most powerful biological antioxidant when it comes to defending cells from hazardous or damaging elements.
Vitamin B5 naturally produces coenzyme A, often known as acetyl-CoA. This coenzyme serves a variety of purposes. It is first and foremost in charge of beginning fatty acid biosynthesis within cells. Fatty acids are essential for life because they produce the phospholipid bilayer that makes up the cell membrane. The citric acid cycle, which results in the generation of ATP, is also started by coenzyme A.
- Non-Vitamins
Non-vitamin coenzymes help enzymes transport chemical information. They ensure that physiological functions in an organism, such as blood coagulation and metabolism, take place. Nucleotides like adenosine, uracil, guanine, and inosine can be used to make these coenzymes.
An example of an essential non-vitamin coenzyme is adenosine triphosphate (ATP). In fact, it is the coenzyme with the greatest distribution in the human body. It carries materials and provides the energy required for chemical reactions and muscular contraction. ATP accomplishes this by transporting both a phosphate and energy to various parts of a cell. The energy is released when the phosphate is removed. The electron transport chain is responsible for this process. There would be little energy available at the cellular level without the coenzyme ATP, and regular living processes would be impossible.
The electron transport chain is depicted in this diagram. NADH, a vitamin-derived coenzyme, kicks things off by delivering electrons. The ultimate product is ATP, which stands for adenosine triphosphate:
2.7.1 Specificity of Enzymes
The specificity of enzymes in relation to the reactions they catalyse is one of the qualities that makes them so valuable as diagnostic and research tools. Only a few enzymes have perfect specificity, which means they will catalyse only one reaction. Other enzymes will have a preference for a specific chemical bond or functional group. There are four different forms of specificity in general:
- Absolute specificity - the enzyme will only catalyse one reaction at a time.
- Group specificity - the enzyme will only work on molecules with certain functional groups, such as amino, phosphate, or methyl.
- Linkage specificity - regardless of the rest of the molecular structure, the enzyme will operate on a certain type of chemical bond.
- Stereochemical specificity - the enzyme will only operate on one steric or optical isomer at a time.
Cofactors may serve a variety of apoenzymes, despite the fact that enzymes have a high degree of specificity. For example, nicotinamide adenine dinucleotide (NAD) is a coenzyme that works as a hydrogen acceptor in a variety of dehydrogenase processes. The alcohol dehydrogenase, malate dehydrogenase, and lactate dehydrogenase processes are among them.
2.7.2 The mechanism of enzymatic action
An enzyme attracts substrates to its active site, catalyzes the chemical reaction by which products are formed, and then allows the products to dissociate (separate from the enzyme surface). The enzyme–substrate complex is the combination of an enzyme and its substrates. The complex is called a ternary complex when two substrates and one enzyme are involved; a binary complex is when one substrate and one enzyme are involved. Electrostatic and hydrophobic forces attract the substrates to the active site, which are referred to as noncovalent bonds since they are physical rather than chemical bonds.
Mechanisms of enzymatic action
Mechanisms of enzymatic action
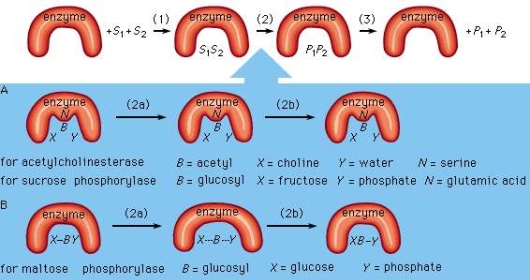
Assume two substrates (S1 and S2) bind to the enzyme's active site in step 1 and then react to create products (P1 and P2) in step 2. In step 3, the products separate from the enzyme surface, allowing the enzyme to be released. The enzyme, which is unaffected by the reaction, is able to generate products by reacting with additional substrate molecules several times per second. The actual chemical transformation phase is of significant interest, and while much is known about it, it is still not completely understood. In general, there are two types of enzymatic mechanisms: those that create a covalent intermediate and those that do not.
One substrate, BX, for example, combines with the group N on the enzyme surface to generate an enzyme-B intermediate compound in the mechanism by which a covalent intermediate—that is, an intermediate with a chemical link between substrate and enzyme—is formed. The intermediate chemical then combines with the second substrate, Y, to produce the BY and X products.
This mechanism is used by many enzymes to catalyse processes. In the sequence that follows, acetylcholinesterase is used as an example. Acetylcholinesterase has two substrates (S1 and S2): acetylcholine (BX) and water (Y). A chemical bond occurs between the acetyl moiety (B) of acetylcholine and the group N (part of the amino acid serine) on the enzyme surface after acetylcholine (BX) binds to the enzyme surface. One product, choline (X), and the enzyme-B intermediate component (an acetyl–enzyme complex) result from the creation of this acyl–serine link. The second product, acetic acid (BY), is formed when the water molecule (Y) combines with the acyl–serine link and dissociates from the enzyme. Acetylcholinesterase is regenerated and can now react with another acetylcholine molecule. A twofold displacement reaction is a type of reaction that involves the production of an intermediate molecule on the enzyme surface.
In a similar way, sucrose phosphorylase works. Sucrose phosphorylase's substrate is sucrose, also known as glucosyl-fructose (BX), and the group N on the enzyme surface is a carboxyl group (COOH). Glucosyl-phosphate (BY) is formed when the enzyme-B intermediate, a glucosyl–carboxyl molecule, combines with phosphate (Y). Fructose is the other product (X).
The covalent intermediate between enzyme and substrate appears to cause the reaction to proceed more quickly in double displacement reactions. Even if the enzyme is momentarily transformed during the enzymatic activity, it nevertheless works as a genuine catalyst at the end of the reaction.
Despite the fact that many enzymes create a covalent intermediate, it is not required for catalysis. In a single displacement reaction, one substrate (Y) reacts directly with the second substrate (XB). The B moiety, which undergoes chemical transformation, is only involved in one reaction and does not make a link with a group on the enzyme surface. For example, the enzyme maltose phosphorylase impacts the bonds between the substrates (BX and X), which in this case are maltose (glucosylglucose) and phosphate, to produce glucose (X) and glucosylphosphate (BY).
Many enzymatic reactions require covalent intermediates between parts of a substrate and an enzyme, and several amino acids—serine, cysteine, lysine, and glutamic acid—are involved.
2.7.3 The Stereochemistry of Enzyme-Catalyzed Reactions:
The stereochemistry ideas we've looked at in this chapter are crucial to the study of living things, despite being difficult to grasp and picture. Chiral centres and/or stereogenic alkene groups are found in the vast majority of biological compounds. Most significantly, proteins are chiral, which includes enzymes that catalyse a cell's chemical reactions, receptors that convey information within or between cells, and antibodies that bind selectively to potentially hazardous intruders. Proteins can recognise and bind to other organic molecules very specifically because they fold up into a certain three-dimensional structure, as you know from your biology lessons. A protein's ligand or substrate could be anything from a simple organic molecule like pyruvate to a huge biopolymer like a specific piece of DNA, RNA, or another protein. Proteins are particularly sensitive to the stereochemistry of their ligands since they are chiral molecules: for example, a protein may bind particularly to (R)-glyceraldehyde but not to (S)-glyceraldehyde, much as your right hand will not fit into a left-handed baseball glove (see end of chapter for a link to an animation illustrating this concept).
Ibuprofen is currently sold as a racemic combination, but only the S enantiomer is useful due to the specific way it binds to and inhibits the action of prostaglandin H2 synthase, an enzyme involved in the body's inflammatory response process.
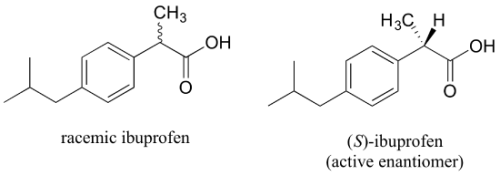
Ibuprofen's R enantiomer does not bind to prostaglandin H2 synthase in the same way that the S enantiomer does, and hence does not have the same inhibitory effect on the enzyme's action. Fortunately, (R)-ibuprofen appears to have no negative side effects and is actually isomerized to (S)-ibuprofen by a bodily enzyme over time.
We examined the tragic case of thalidomide earlier in this chapter, and how it appears that the S enantiomer is the one that causes birth abnormalities. Many various theories have been proposed over the years to explain the drug's teratogenic (birth defect-causing) impact, but the scientific community has yet to reach a consensus. However, in 2010, a group of researchers in Japan discovered that thalidomide attaches specifically to a protein known as 'thereblon.' Furthermore, when thereblon production is inhibited in female zebra fish, developmental problems in her offspring arise that are strikingly similar to those induced by thalidomide treatment, implying that thalidomide binding inactivates the protein, resulting in the teratogenic impact.
You can directly experience the biological importance of stereoisomerism by going to the grocery shop. Carvone is a chiral, plant-derived chemical that contributes to the aromas of spearmint and caraway (a spice) in their R and S forms, respectively.
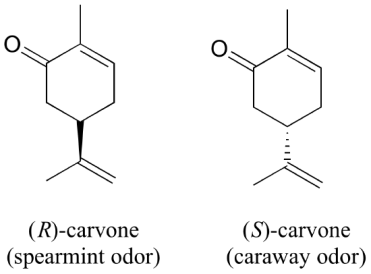
Although details are unknown, the two enantiomers are thought to interact differently with one or more smell receptor proteins in your nose, resulting in different chemical signals being transmitted to your brain's olfactory region.
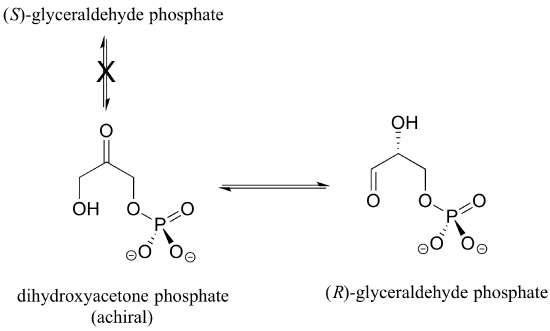
When it comes to the stereochemistry of the reactions that enzymes catalyse, they are exceedingly particular. With very few exceptions, when the product of a biological reaction contains a chiral centre or a stereogenic alkene, only one stereoisomer of the product is generated. The enzyme triose-phosphate isomerase, for example, catalyses the reversible interconversion of dihydroxyacetone (which is achiral) and (R)-glyceraldehyde phosphate in the glycolysis pathway. This enzyme does not produce the (S)-glyceraldehyde enantiomer in the left-to-right reaction, and it is not employed as a starting molecule in the right-to-left process because it does not 'fit' in the enzyme's active site.
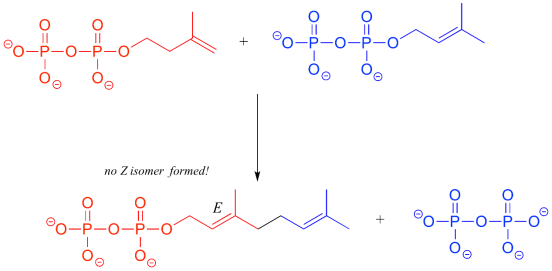
Two five-carbon building-block molecules combine to generate a ten-carbon chain with an E-alkene group in the isoprenoid biosynthesis pathway. The production of the Z diastereomer is not catalysed by the enzyme.
2.8.1 Definition, classification, and main properties
Enzymes are a collection of chemical molecules that are grouped together because they all have one thing in common: they can inhibit enzyme function. The inhibition of activity is caused by the inhibitor attaching to the enzyme molecule, which stops the catalytic reaction. Enzyme inhibitors are significant in the development of various sciences (biochemistry, physiology, pharmacy, agriculture, ecology) and technologies since enzymes catalyse the majority of chemical reactions in living organisms (production of pharmaceutical drugs, insecticides, pesticides, chemical weapons, etc.).
Enzyme inhibitors are found in several pharmaceuticals. Nonsteroidal anti-inflammatory drugs (NSAIDs) are inhibitors of the enzyme cyclooxygenase, which catalyses the first step in the synthesis of biologically active compounds prostaglandins, which are responsible for the development of pain, inflammation, fever, smooth muscle contraction, blood clot formation, and other conditions.
All inhibitors may be combined in different groups in accordance with their chemical structure: ions of metals (Hg+, Fe2+, Cu+, Pb2+), organic compounds (e.g., N-ethylmaleimide, diisopropyl phosphofluoridate, oligomycin), and large bioorganic molecules, (peptides, proteins, etc). However, this classification does not reflect mechanism of their interaction with enzyme.
Enzyme inhibitors are classified into two categories based on their manner of action (reversible and irreversible inhibitors). Reversible inhibitors, on the other hand, can be divided into four classes based on their kinetic behaviour (competitive, uncompetitive, non-competitive, and mixed inhibitors).
The creation of an enzyme-inhibitor complex (EI complex) with no (or poor) enzyme activity is part of the mechanism of action of enzyme inhibitors. Because it is strongly attached to the enzyme, an irreversible inhibitor dissociates from this complex very slowly. The creation of a covalent link or a hydrophobic contact between the enzyme and the inhibitor is primarily responsible for this way of inhibition. In most cases, irreversible inhibitors react with the enzyme and chemically alter it. These inhibitors frequently contain reactive functional groups that change critical amino acid residues in enzymes. They can also provide inhibition by altering the structure of the enzyme. N-ethylmaleimide, for example, is an irreversible inhibitor that covalently interacts with the SH-group of cysteine residues in enzyme molecules such as peptidase (insulin-degrading enzyme), 3-phosphoglyceraldehyde dehydrogenase, or a hydrophobic compound from a group of cardiotonic steroids that, in the end, binds to Na, K-ATPase via hydrophobic interactions. Another well-known irreversible inhibitor is diisopropyl phosphofluoridate, which is a strong neurotoxin that changes the OH-group of serine residues in the active site of enzymes like chymotrypsin and other serine proteases or acetylcholine esterase in cholinergic synapsis of the nervous system. When this enzyme is inhibited, the concentration of the neurotransmitter acetylcholine rises, resulting in muscle paralysis and death. Aspirin (acetyl salicylic acid), a cyclooxygenase inhibitor, covalently changes the OH-group of a serine residue near the cyclooxygenase active site.
Irreversible inhibition is not the same as irreversible inactivation of an enzyme. Irreversible inhibitors are enzyme inhibitors that are unique to a single enzyme class and do not inactivate all proteins. Detergents, unlike denaturers like urea, do not damage protein structure; instead, they change the active site of the target enzyme.
As a result of the tight binding, removing an irreversible inhibitor from the EI complex once it has formed is challenging. So, if after the formation of the EI complex, dilution with a substantial volume of water (100–200 excess) does not restore enzyme activity, we can designate to a chemical molecule as an irreversible enzyme inhibitor.
Time-dependent reduction of enzyme activity is seen with irreversible inhibitors. Irreversible inhibitor-enzyme interaction is a bimolecular reaction:
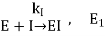
Where E is the enzyme, I is the inhibitor, EI is the enzyme-inhibitor complex, and ki is the reaction velocity constant.
However, when the concentration of the inhibitor is much higher than the concentration of the enzyme, the activity of irreversible inhibitors is usually characterised by the constant of observed pseudo-first order process. Plotting the ln of enzyme activity (in percent relative enzyme activity in the absence of inhibitor) vs. Time can be used to determine the value of pseudo-first order rate of inhibition. The tangent of the slope angle of a straight line determined in this manner equals the value of the pseudo-first order inhibition constant. By dividing the acquired value of pseudo-first order reaction constant per inhibitor concentration, the value of rate constant of bimolecular reaction for irreversible inhibition may be derived.
Reversible inhibitors bind to enzymes in a reversible manner. It means that the formation and dissociation of the EI complex are in balance:
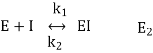
Where k1 is a constant of the velocity of direct reaction and k2 is a constant of the velocity of reverse reaction. The effect of reversible inhibitors is characterized by the constant of dissociation of EI complex that is equal to [E] [I]/[EI] or k1/k2.
Non-covalent interactions like as hydrogen or ionic bonds are usually used by reversible inhibitors to bind to enzymes. Depending on whether these inhibitors bind to the enzyme, the enzyme-substrate complex, or both, different types of reversible inhibition are produced.
Competitive inhibition is one type of reversible inhibition. There are two sorts of complexes in this case: enzyme inhibitor (EI) and enzyme substrate (ES); EI has no enzyme activity. The enzyme cannot bind to both the substrate and the inhibitor at the same time. Increases in substrate concentration can counteract this inhibition. The maximum velocity (Vmax) remains constant, though. The apparent Km value will grow, while the maximum velocity (Vmax) will remain unchanged (Figure). Competitive inhibition can occur not just in respect to substrate, but also in connection to cofactors and activators.
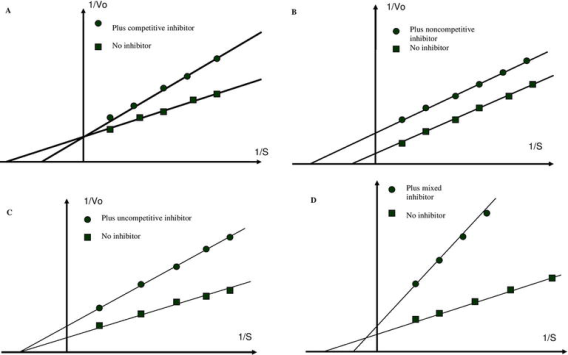
Kinetic test for reversible inhibitor classification. Double reciprocal plot (1/Vo) vs. (1/s) for competitive (A), uncompetitive (B), non-competitive (C), and mixed (D) enzyme inhibition.
Uncompetitive inhibition is another type of reversible inhibition. The inhibitor binds just to the substrate-enzyme complex in this situation; it does not interfere with substrate binding to the active site but prevents the dissociation of the complex enzyme substrate, resulting in inhibition being solely dependent on inhibitor concentration and Ki value. This sort of inhibition causes Vmax and Km to fall (Figure B).
Non-competitive inhibition is the third form of inhibition. This form of inhibition prevents the complex enzyme (E) inhibitor (I) substrate (EIS) from dissociating and producing a reaction product. The inhibitor binds to the E or ES complex in this situation. The inhibitor's attachment to the enzyme decreases its activity but has no effect on substrate binding. As a result, the extent of inhibition is solely determined by the inhibitor's concentration. Vmax will fall in this instance, but Km will remain unchanged (Figure C).
Mixed inhibition occurs when the inhibitor can bind to the enzyme and the enzyme-substrate complex at the same time. The inhibitor's binding, on the other hand, has an effect on the substrate's binding and vice versa. Increased substrate concentrations can diminish, but not eliminate, this sort of inhibition. Although mixed-type inhibitors can bind to the active site, this inhibition is usually due to the inhibitor's allosteric action (see below). This type of inhibitor will reduce Vmax while increasing Km (Figure C).
Inhibition by excess substrate or product is a special example of enzyme inhibition. This inhibition can take one of three forms: competitive, non-competitive, or mixed. When a dead-end enzyme-substrate complex forms, the enzyme is inhibited by its substrate. In many cases of substrate inhibition, a molecule of substrate binds to the active site in two places (for example, at the “head” and “tail” of the molecule). At high concentrations, two substrate molecules bind in the active site in the following way: one molecule connects with the "head," while the other molecule binds with the "tail." This binding is ineffective, as the substrate cannot be transformed into the desired product. The inhibition of acetyl cholinesterase by an excess of acetylcholine is an example of this type of inhibition.
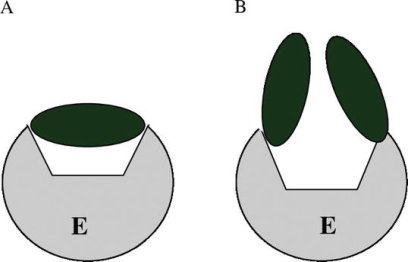
Substrate inhibition of enzymes. Unproductive binding of two substrate molecules with the same site (A) and productive binding of one substrate molecule with two points of enzyme active site (B) (B).
Competitive inhibitors primarily interact with the active site of the enzyme, inhibiting genuine substrate binding. The suppression of fumarate hydratase by maleate, a substrate homolog, is a classic example of competitive inhibition. The hydration of the trans-double bound of fumarate is catalysed by the enzyme, but not by maleate (cis-isomer of fumarate). Maleate binds to the active site with a high affinity, preventing fumarate from binding. Maleate cannot be transformed into the reaction product despite binding to the active site. Maleate, on the other hand, binds to the active site, making it inaccessible to the actual substrate and so inhibiting it.
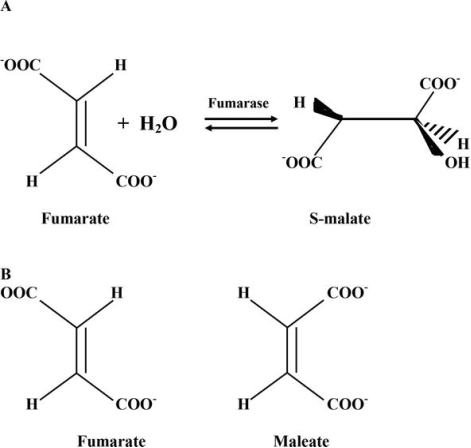
Competitive inhibitors of enzymes are an example. A reaction catalysed by fumarate hydratase (A) and a structural comparison of fumarate (reaction substrate) and maleate (competitive inhibitor of enzyme) (B).
Some reversible inhibitors adhere to the enzyme so tightly that they become irreversible. Proteolytic enzymes from the gastrointestinal tract are known to be released inactively by the pancreas. Proenzyme digestion with limited trypsin is used to activate them. The pancreas generates trypsin inhibitor to prevent proteolytic enzyme activity. It's a tiny molecule of protein (it consists of 58 amino acid residues). With a Kd of 0.1 pM, this inhibitor binds directly to the active site of trypsin. Complex EI does not dissolve even in a solution of 6 M urea, indicating that the binding is nearly irreversible. The inhibitor is a very efficient mimic of trypsin substrates; the inhibitor's Lys-15 amino acid binds with an aspartic residue on the enzyme's surface that is slated for substrate binding, inhibiting substrate binding and conversion into the product.
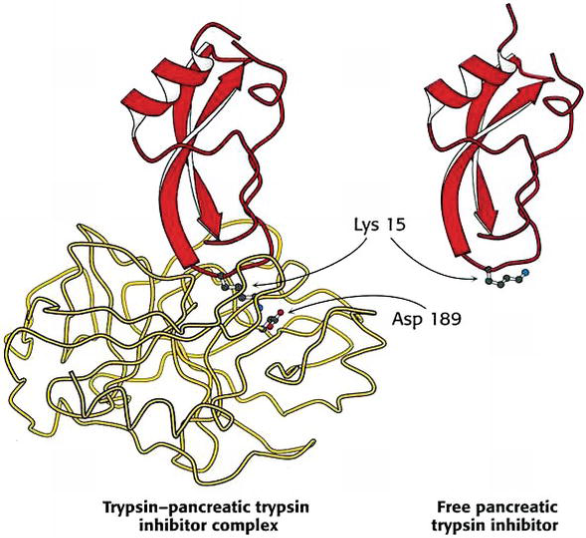
Structure of complex pancreatic trypsin inhibitor—trypsin and free trypsin inhibitor.
2.8.2 Irreversible inhibitors as a tool for study of enzymes: enzyme active sites labeling by irreversible inhibitors
To learn more about how enzymes work, we need to figure out which functional groups are essential for enzyme activity and where they are positioned in the active site. The first method is to use X-ray crystallography to reveal the 3D structure of the enzyme with bound substrate. A different and/or supplementary strategy is to utilise a group-specific reagent that is also an irreversible enzyme inhibitor. It has the ability to covalently attach to reactive groups in the active site of enzymes, allowing for the elucidation of functional amino acid residues. After complete enzyme inhibition, enzyme proteolysis, and identification of labelled peptides, modified amino acid residues may be discovered (s).
Irreversible inhibitors for this purpose can be separated into two categories: (1) group-specific reagents for reactive chemical groups and (2) substrate analogues with functional groups that can interact with reactive amino acid residues. These chemicals can covalently alter crucial amino acids for enzyme active site activity and identify them in this way.
Diisopropyl phosphofluoridate was one of the most well-known group-specific reagents used to identify functional amino acid residues of the enzyme active site of the protease chymotrypsin. It only changed one of the enzyme's 28 serine residues. This indicates that the serine residue is quite reactive. The presence of Ser-195 in the active site of chymotrypsin was confirmed later in an analysis, and the source of its high reactivity was discovered. Diisopropyl phosphofluoridate was also utilised to successfully identify a reactive serine residue in acetylcholinesterase's active site.
Several SH-reagents, including C-labeled N-ethylmaleimide, iodoacetate, and iodoacetamide, were employed to show reactive SH-groups in the active sites of various enzymes. Cysteine was discovered in the active sites of several dehydrogenases, cysteine proteases, and other enzymes using these reagents.
The use of reactive substrate analogues is the second method. Chemically reactive groups can covalently attach to some amino acid residues in these molecules, which are structurally identical to the substrate. Substrate analogues are more specific than reagents that target a specific group. The identification of Hys-57 in the chymotrypsin active site was made feasible by tosyl-L-phenylalanine chloromethyl ketone, a substrate analogue for chymotrypsin that can bind covalently with histidine residue and inhibit enzyme irreversibly.
2.8.3 Natural enzyme inhibitors
Proteins or peptides that directly bind to and inhibit target enzymes make up the majority of cellular enzyme inhibitors. These particular chemicals that are generated in organisms affect a variety of metabolic pathways. Protein serpins are an excellent example of these inhibitors. It's a huge protein family with comparable structures. The majority of them are chymotrypsin-like serine protease inhibitors.
Serine proteases (e.g., chymotrypsin) feature a reactive serine residue in their active site and comparable catalytic processes. The peptide bond is cleaved in two steps by these proteases. In the beginning of the catalytic act, a reactive serine residue in the protease active site loses H+ and becomes nucleophilic, attacking the substrate peptide bond. This causes the enzyme to release a new N-terminal section of the protein substrate (first product) and establish a covalent ester bond with the second component of the substrate. The hydrolysis of the ester bond and the release of the second product are the results of the second phase of catalysis of common substrates (C-terminal part of protein substrate). If a serine protease cleaves serpin, it goes through a conformational change before the ester link between the enzyme and the second half of the substrate is hydrolyzed (serpin). The “freezing” of intermediates is caused by a change in serpin conformation (complex of enzyme with covalently attached second part of serpin is retained for several days). Serpins are therefore irreversible inhibitors with a peculiar mode of action. Because each serpin molecule may inactivate a single molecule of protease and kills itself during the process of protease inhibition, they've given them the label "suicide inhibitors."
When considering enzyme inhibitors, it's important to remember that many living organisms are engaged in a "chemical battle." Antibiotics are being used by fungi to battle bacteria for nourishment. Poisons are used by most stationary creatures, such as plants and some sea invertebrates, to defend themselves from being eaten; however, some vertebrates (such as snakes) and invertebrates (such as bees and wasps) utilise poisons not just for defence but also for food. If we look at the chemistry of these toxins, we can see that they contain a variety of enzyme inhibitors. They were chosen through evolution to inhibit various metabolic processes that contribute to death in victims' organisms.
Plant and invertebrate poisons have been utilised as medicine for thousands of years. However, it was not until the twentieth century that it was discovered that the poisons contain a variety of enzyme inhibitors as well as blockers of other biological components (channels, receptors, etc.) Bee venom, for example, contains melittin, a 28-amino-acid peptide. This peptide can interact with a variety of enzymes, inhibiting their activity; in particular, it binds to the protein calmodulin, which is an enzyme activator. Special studies have revealed that the structure of melittin is similar to that of some proteins (or, more precisely, some parts of protein molecules) that interact with the target enzyme to provide biological function.
Another example of natural inhibitors is cardiotonic steroids that were found initially in plants (digoxin, digitonin, ouabain) and in the mucus of toads (marinobufagenin, bufotoxin, etc.). These compounds are irreversible inhibitors of Na, K-ATPase that is enzyme transporting Na+ and K+ through the plasma membrane of animals against the electrochemical gradients. In the end of the twentieth century, it was shown that cardiotonic steroids are presented in low concentrations in the blood of mammals including human beings. The increase of concentration of these compounds in the blood may be involved in the development of several cardiovascular and renal diseases including volume-expanded hypertension, chronic renal failure, and congestive heart failure.
Natural poisons are a strong tool for studying enzyme activity, and a thorough understanding of how they work is required for these investigations. It could also serve as a model for developing new inhibitors and activators that mimic natural chemicals with similar characteristics.
2.8.4 Enzyme inhibitors as pharmaceutical agents
Nonsteroidal anti-inflammatory medications (NSAIDs) that inhibit cyclooxygenase have already been mentioned. This class of substances (the most commonly prescribed medications in the world, the oldest of which is aspirin) has been successfully used for more than a century to treat people with fever, cardiovascular disease, joint pain, and other ailments all over the world. Irreversible and reversible inhibitors of prostaglandin synthesis, which affect many aspects of inflammation, smooth muscle contraction, and blood clotting, are among these medications. However, there are many additional types of medications that are enzyme inhibitors by nature; the following enzyme inhibitors are currently being developed by pharmaceutical companies and have significant therapeutic implications.
Angiotensin-converting enzyme inhibitors (ACE). By removing a dipeptide from angiotensin I's C-terminus, ACE catalyses the conversion of inactive decapeptide angiotensin I into angiotensin II. The vasoconstrictor angiotensin II is very strong. The inhibition of ACE causes the concentration of angiotensin I to drop and the smooth muscles of the arteries to relax. Drugs that inhibit ACE are commonly used to treat arterial hypertension.
Inhibitors of the proton pump (PPIs). The proton pump is an enzyme found in the parietal cells of the stomach mucosa's plasma membrane. It's a P-type ATPase that uses the energy of ATP cleavage to deliver proton secretion from parietal cells in the gastric cavity against an electrochemical gradient. PPIs are a class of substituted benzopyridines that are transformed into active sulfonamides in the stomach's acid medium by interacting with cysteine residues in the pump. As a result, PPIs are acid-activated prodrugs that are transformed into drugs within the body. In the 1980s of the twentieth century, PPIs were first used in clinical practise. The medications have been successfully used to treat gastritis, gastric and duodenal ulcers, and gastroesophageal reflux disease since that period.
Statins represent a group of compounds that are analogs of mevalonic acid. They are inhibitors of 3-hydroxy-3-methylglutaryl-CoA reductase, an enzyme participating in cholesterol synthesis. Statins are used as drugs preventing or slowing the development of atherosclerosis. Because of the existence of some adverse effects, statins may be recommended for patients that cannot achieve a decrease of cholesterol level in the blood through diet and changes in lifestyle.
Penicillin, an antibiotic, covalently alters the enzyme transpeptidase, blocking bacterial cell wall formation and ultimately killing the germs.
Methotrexate is a structural homologue of tetrahydrofolate, a cofactor for the enzyme dihydrofolate reductase, which catalyses an essential step in purine and pyrimidine production. Methotrexate inhibits nucleotide base synthesis by binding to this enzyme 1000 times more firmly than the substrate. It's utilised in cancer treatment.
A promising new anticancer therapy that is linked to the regulation of protein kinases that control the cellular response to DNA damage is now in the research stage. Selective inhibitors of these enzymes are now being studied in cancer patients in clinical trials.
Recently, two different types of enzyme inhibitors were used to produce a breakthrough in the treatment of people with acquired immune deficiency syndrome (AIDS) caused by the human immunodeficiency virus (HIV). Patients with this disease should now be treated with nucleoside reverse transcriptase inhibitors and protease inhibitors. These inhibitors have also been shown to have anticancer properties and are effective against a variety of different viral infections. This page is a partial list of enzyme inhibitors that are used in the treatment of a variety of diseases. However, the points listed above indicate how beneficial and crucial therapeutic use of theoretical knowledge gained from enzyme inhibitor research is.
New inhibitors of known enzymes with therapeutic potential are being sought by scientists all over the world. A project devoted to the discovery, manufacture, and analysis of new inhibitors of carbonic anhydrase, an enzyme involved in the development of symptoms and diseases such as edoema, glaucoma, obesity, cancer, epilepsy, and osteoporosis, is an example of this difficult research.
2.9.1 Routes of exposure and absorption of chemicals
Injection
Because the chemical is injected directly into the body, injection is the sole form of exposure for poisons in which the complete amount exposed is absorbed independent of the chemical administered. Chemicals can be injected intravenously (directly into a vein), intramuscularly (straight into a muscle), subcutaneously (under the skin), or intraperitoneally (straight into the abdominal cavity) (within the membrane lining the organs of the abdomen).
Intravenous injection is the fastest way to introduce a chemical into the body since blood is the vehicle for chemical distribution in the body. Because of the near-instantaneous dispersion and irreversibility, intravenous injection is a risky type of chemical exposure, with a good probability of producing drug overdose if done incorrectly.
Chemicals are taken into the bloodstream rather quickly following intramuscular injection due to the substantial blood supply to the skeletal muscles. The low blood flow in the subcutaneous tissues is likely to blame for the sluggish absorption of a drug into the blood following a subcutaneous injection. Only biomedical research employs intraperitoneal injection. Because of the abundant blood flow to the belly, absorption is rather quick with intraperitoneal injection.
Ingestion
The most common way to be exposed to harmful substances is by ingestion. The degree to which a chemical is ionised is significant in deciding whether it is absorbed because most compounds diffuse over the cell membrane in their nonionized state (see above Transport of chemicals through a cell membrane).
When the pH of the environment changes, organic acids and bases dissociate into their ionised forms. In an acidic environment (such as the stomach), organic acids are nonionized and so diffuse over a membrane, whereas organic bases are nonionized and hence diffuse across a membrane in a basic environment (such as in the intestine).
Chemical absorbabilities of distinct segments of the gastrointestinal system differ due to differences in pH and surface area. The stomach and small intestine are the primary sites of poison absorption, with the latter accounting for the majority of the absorption. The intestine has a larger surface area and a better blood supply. Numerous projections on the luminal surface are housed in folds in the small intestine mucosa, increasing the surface area of the 280-centimetre-long (110-inch-long) small intestine to up to 2,000,000 square centimetres.
The small intestine's mucosal surface has an alkaline pH. Organic bases are generally absorbed in their nonionized, lipid-soluble form. The stomach contents have a pH of 1 to 2 (highly acidic), and mild organic acids are typically nonionized and lipid-soluble. The poisons should be absorbed there, but because the stomach's surface area is much smaller than the small intestine's, the contents of the stomach (together with the poisons) are frequently transmitted to the intestine before the chemicals are absorbed. The low absorption of organic bases by the stomach is mostly due to the stomach's acidic environment.
2.9.2 Enzyme Inhibition
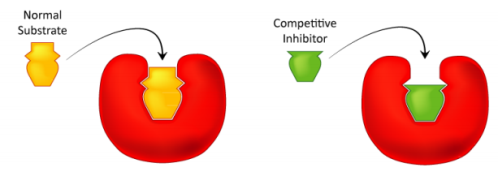
Figure: Line-Weaver Burk Plot of competitive inhibition
2.9.3 No Effect On VMAX
What method do we use to investigate competitive inhibition? This is how it's usually done. First, a set of V vs. [S] reactions is carried out without the use of an inhibitor (20 or so tubes, with buffer and constant amounts of enzyme, varying amounts of substrate, equal reaction times). If desired, V vs. [S] and 1/V vs. 1/[S] are plotted. The second set of reactions is carried out in the same way as the first, except that each tube contains a predetermined dose of methotrexate inhibitor. The inhibitor successfully competes for the enzyme at low substrate concentrations, but at high substrate concentrations, the inhibitor has a significantly diminished effect since the substrate outcompetes it due to its larger concentration (remember that the inhibitor is at fixed concentration). The outcomes of these tests are depicted graphically above. The competitive inhibitor has virtually no effect at high substrate concentrations, causing the enzyme's Vmax to stay constant. To clarify, this is owing to the inhibitor's inability to compete effectively at high substrate concentrations. It does, however, at lower substrate concentrations.
What method do we use to investigate competitive inhibition? This is how it's usually done. First, a set of V vs. [S] reactions is carried out without the use of an inhibitor (20 or so tubes, with buffer and constant amounts of enzyme, varying amounts of substrate, equal reaction times). If desired, V vs. [S] and 1/V vs. 1/[S] are plotted. The second set of reactions is carried out in the same way as the first, except that each tube contains a predetermined dose of methotrexate inhibitor. The inhibitor successfully competes for the enzyme at low substrate concentrations, but at high substrate concentrations, the inhibitor has a significantly diminished effect since the substrate outcompetes it due to its larger concentration (remember that the inhibitor is at fixed concentration). The outcomes of these tests are depicted graphically above. The competitive inhibitor has virtually no effect at high substrate concentrations, causing the enzyme's Vmax to stay constant. To clarify, this is owing to the inhibitor's inability to compete effectively at high substrate concentrations. It does, however, at lower substrate concentrations.
2.9.4 Increased KM
When the inhibitor is present, the apparent KM of the enzyme for the substrate increases (-1/KM gets closer to zero - red line above), demonstrating the inhibitor's improved competitiveness at lower substrate concentrations. It may not be clear why we refer to the modified KM as the enzyme's apparent KM. The reason for this is that the inhibitor has no effect on the enzyme's affinity for folate as a substrate. It merely appears to be that way. Because of how competitive inhibition works, this is the case. The enzyme is effectively "put out of operation" when the competitive inhibitor binds to it. Inactive enzymes have neither a substrate affinity nor an activity. We can't calculate KM for an enzyme that isn't active.
The enzyme molecules that aren't affected by methotrexate can still bind folate and function. They are unaffected by methotrexate, and their KM values remain unaltered. Why, then, does KM appear to be higher when a competitive inhibitor is present? The reason for this is because the competitive inhibitor reduces the amount of active enzyme at low substrate concentrations. When the amount of enzyme is lowered, more substrate is required to provide the reduced amount of enzyme with enough energy to reach Vmax/2.
It's worth mentioning that the proportion of inactive enzyme under competitive inhibition varies dramatically depending on the [S] values employed. To begin, the enzyme is inhibited to the greatest extent at low [S] values. No significant percentage of enzyme is inhibited at high [S]. As we'll see in non-competitive inhibition, this isn't always the case.
2.9.5 Non-Competitive Inhibition
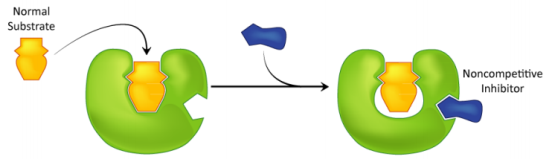
Figure: Penicillin
2.10.1 Nucleic Acids:
Nucleic acid is a naturally occurring chemical molecule that can be broken down to produce phosphoric acid, sugars, and an organic base combination (purines and pyrimidines). Nucleic acids are the cell's principal information-carrying molecules, and they determine every living thing's inherited feature by directing the process of protein synthesis. Deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) are the two main types of nucleic acids (RNA). The genetic material in all free-living organisms and most viruses is made up of DNA, which is the master blueprint for life. RNA is the genetic material of some viruses, but it may also be present in all living cells, where it plays a key part in processes like protein synthesis.
This article delves into the chemistry of nucleic acids, outlining the structures and properties that enable them to act as genetic information transmitters. See heredity for further information on the genetic code, and metabolism for more information on the role of nucleic acids in protein synthesis.
2.10.2 Nucleotides: building blocks of nucleic acids
Basic structure
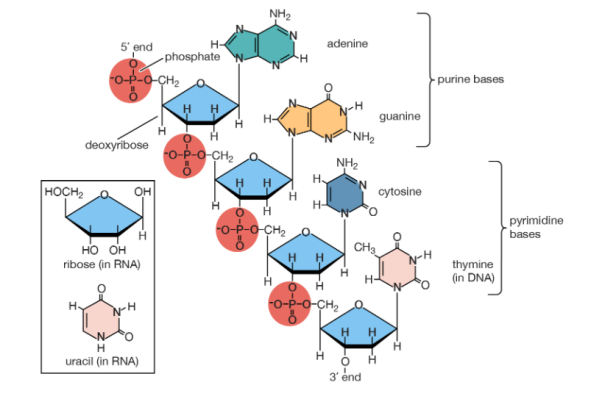
Polynucleotides, or long chainlike molecules made up of a number of almost similar building units called nucleotides, make up nucleic acids. A nitrogen-containing aromatic base is connected to a pentose (five-carbon) sugar, which is then connected to a phosphate group in each nucleotide. Adenine (A), guanine (G), cytosine (C), thymine (T), and uracil (U) are the four nitrogen-containing bases found in each nucleic acid (U).
Purines A and G are purines, while pyrimidines C, T, and U are pyrimidines. The nucleotides A, C, and G are found in all nucleic acids; T, on the other hand, is only found in DNA, while U is only found in RNA. The pentose sugar in DNA (2′-deoxyribose) differs from the sugar in RNA (ribose) by the absence of a hydroxyl group (―OH) on the 2′ carbon of the sugar ring. A nucleoside is a sugar connected to one of the bases that does not have a phosphate group linked to it. By bridging the 5′-hydroxyl group on one sugar to the 3′-hydroxyl group on the next sugar in the chain, the phosphate group joins successive sugar residues. Phosphodiester bonds are nucleoside connections that are found in both RNA and DNA.
Biosynthesis and degradation
Nucleotides are made in the cell from easily available precursors. The pentose phosphate pathway is used to make the ribose phosphate part of both purine and pyrimidine nucleotides. The six-atom pyrimidine ring is first formed, then linked to the ribose phosphate. Purines' two rings are formed as adenine or guanine nucleosides are assembled, while coupled to the ribose phosphate. The end product in both circumstances is a nucleotide with a phosphate bonded to the sugar's 5′ carbon. Finally, a kinase enzyme adds two phosphate groups to ribonucleoside triphosphate, the immediate precursor of RNA, utilising adenosine triphosphate (ATP) as the phosphate donor. To make deoxyribonucleoside diphosphate, the 2′-hydroxyl group is removed from ribonucleoside diphosphate. Another kinase then adds another phosphate group from ATP to make deoxyribonucleoside triphosphate, the immediate precursor of DNA.
RNA is regularly generated and destroyed down during regular cell metabolism. Several salvage processes reuse purine and pyrimidine residues to create additional genetic material. Purine is recovered as the matching nucleotide, whereas pyrimidine is recovered as the nucleoside.
2.10.3 Roles of DNA and RNA in cells
Nucleic acids, which are macromolecules made up of nucleotide units, come in two types: deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) (RNA). DNA is the genetic material that can be found in all living things, from single-celled bacteria to multicellular animals like you and me. Although some viruses employ RNA rather than DNA as their genetic material, they aren't really alive (since they cannot reproduce without help from a host).
2.10.4 DNA in cells
DNA is present in the nucleus, a specialised, membrane-bound vault in the cell, as well as in some other types of organelles in eukaryotes, such as plants and animals (such as mitochondria and the chloroplasts of plants). Although DNA is located in a specialised cell area termed the nucleoid in prokaryotes, such as bacteria, it is not covered in a membrane envelope.
DNA is often divided up into a number of very long, linear segments called chromosomes in eukaryotes, but chromosomes in prokaryotes, such as bacteria, are much smaller and often circular (ring-shaped). A chromosome can have tens of thousands of genes, each of which gives instructions on how to manufacture a specific product that the cell requires.
From DNA to RNA to proteins
Many genes code for protein products, which means they specify the amino acid sequence utilised to make a certain protein. However, before this information can be used to make proteins, an RNA copy (transcript) of the gene must be created. Messenger RNA (mRNA) is a form of RNA that acts as a link between DNA and ribosomes, which are molecular engines that read mRNA sequences and use them to make proteins. The "core dogma" of molecular biology is the pathway from DNA to RNA to protein.
Not all genes produce protein products, which is important to note. Some genes, for example, code for ribosomal RNAs (rRNAs), which are structural components of ribosomes, or transfer RNAs (tRNAs), which are cloverleaf-shaped RNA molecules that transport amino acids to the ribosome for protein synthesis. Other RNA molecules, such as small microRNAs (miRNAs), operate as gene regulators, and new non-protein-coding RNAs are being discovered all the time.
Nitrogenous bases
Organic (carbon-based) compounds with nitrogen-containing ring structures make up the nitrogenous bases of nucleotides.
Adenine (A), guanine (G), cytosine (C), and thymine (T) are the four nitrogenous bases that can be found in DNA (T). Purines, such as adenine and guanine, have two joined carbon-nitrogen rings in their architecture. In contrast, cytosine and thymine are pyrimidines with a single carbon-nitrogen ring. Adenine, guanine, and cytosine bases can also be found in RNA nucleotides, but instead of thymine, they have uracil, a pyrimidine base (U). Each base has its own structure, as illustrated in the diagram, with its own collection of functional groups connected to the ring structure.
The nitrogenous bases are commonly referred to by their one-letter symbols, A, T, G, C, and U, in molecular biology shorthand. A, T, G, and C are found in DNA, while A, U, G, and C are found in RNA (that is, U is swapped in for T).
Sugars
DNA and RNA nucleotides have somewhat distinct sets of bases, as well as slightly different sugars. Deoxyribose is the five-carbon sugar in DNA, while ribose is the sugar in RNA. These two have relatively similar structures, with one exception: ribose's second carbon has a hydroxyl group, whereas deoxyribose's comparable carbon has a hydrogen. As illustrated in the image above, the carbon atoms of a nucleotide's sugar molecule are numbered 1′, 2′, 3′, 4′, and 5′ (1′ means "one prime"). The sugar is in the middle of the nucleotide, with the base connected to its 1′ carbon and the phosphate group (or groups) to its 5′ carbon.
Phosphate
A single phosphate group, or a chain of up to three phosphate groups, can be added to the sugar's 5' carbon in nucleotides. Although some chemistry sources limit the term "nucleotide" to the single-phosphate case, the broader meaning is widely recognised in molecular biology. 11superscript begin, 1 superscript end
A nucleotide nearing the end of a polynucleotide chain in a cell will have a succession of three phosphate groups. The nucleotide loses two phosphate groups when it joins the developing DNA or RNA chain. Each nucleotide in a DNA or RNA strand has only one phosphate group.
2.10.5 Polynucleotide chains
A polynucleotide chain with directionality – that is, two ends that are distinct from each other – is a result of the structure of nucleotides. The 5' phosphate group of the first nucleotide in the chain sticks out at the 5' end, or commencement, of the chain. The 3' hydroxyl of the previous nucleotide added to the chain is exposed at the other end, known as the 3' end. DNA sequences are commonly written in the 5' to 3' manner, with the 5' end nucleotide coming first and the 3' end nucleotide coming last.
A strand of DNA or RNA expands at its 3' end as additional nucleotides are introduced, with the 5′ phosphate of an incoming nucleotide binding to the hydroxyl group at the 3' end of the chain. This forms a chain, with each sugar connected to its neighbours by a series of links known as phosphodiester linkages.
2.10.6 Properties of DNA
Chains of deoxyribonucleic acid, or DNA, are usually found in a double helix, which is a configuration in which two complementary (matching) chains are locked together, as seen in the diagram at left. The sugars and phosphates on the exterior of the helix make up the backbone of DNA, which is frequently referred to as the sugar-phosphate backbone. The nitrogenous bases extend into the interior in pairs, like the steps of a staircase; each pair's bases are linked by hydrogen bonds.
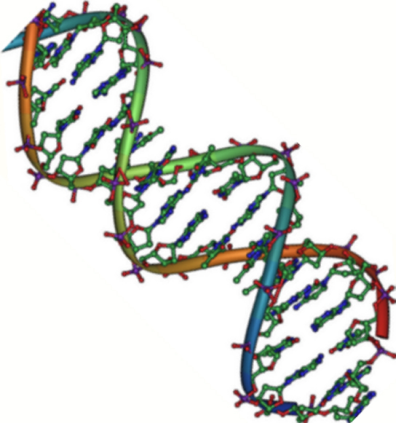
A DNA double helix structural model
The helix's two strands flow in opposing directions, so one strand's 5′ end is hooked up with the 3′ end of its matching strand. (This is known as antiparallel orientation, and it is critical for DNA copying.)
So, can any two bases decide to join forces and form a double helix pair? The answer is emphatically no. Base pairing is highly particular due to the sizes and functional groups of the bases: A can only pair with T, and G can only pair with C, as illustrated below. This means that the interaction between the two strands of a DNA double helix is fairly predictable.
For example, if you know one strand's sequence is 5'-AATTGGCC-3', the complementary strand's sequence must be 3'-TTAACCGG-5'. As a result, each base can be matched with its partner:
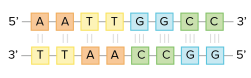
Each base in one strand adheres to its counterpart in the other. Two hydrogen bonds join the A-T pairs, whereas three hydrogen bonds connect the G-C pairs.
When two DNA sequences match in this way, such that they can stick to each other in an antiparallel fashion and form a helix, they are said to be complementary.
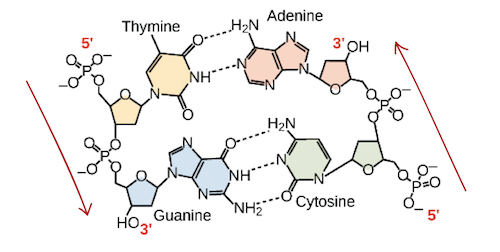
In a double helix of antiparallel strands, hydrogen bonding between complimentary bases maintains DNA strands together. Thymine and adenine make two hydrogen bonds, while guanine and cytosine make three hydrogen bonds.
2.10.7 Properties of RNA
Unlike DNA, ribonucleic acid (RNA) is normally single-stranded. Ribose (the five-carbon sugar), one of the four nitrogenous bases (A, U, G, or C), and a phosphate group make up an RNA nucleotide. We'll look at messenger RNA (mRNA), ribosomal RNA (rRNA), transfer RNA (tRNA), and regulatory RNAs, which are the four major forms of RNA.
Messenger RNA (mRNA)
A protein-coding gene and its protein output are separated by messenger RNA (mRNA). When a cell needs to generate a specific protein, the gene encoding that protein is turned "on," which means an RNA-polymerizing enzyme is summoned to create an RNA copy of the gene's DNA sequence, known as a transcript. The transcript has the same information as the gene's DNA sequence. In the RNA molecule, however, the letter T is substituted with the nucleotide U. For instance, if a DNA coding strand has the sequence 5’-AATTGCGC-3’, the sequence of the corresponding RNA will be 5’-AAUUGCGC-3’.
An mRNA will associate with a ribosome, a molecular mechanism that specialises in constructing proteins from amino acids, once it has been created. The ribosome “reads out” the mRNA nucleotides in groups of three (called codons) and adds a specific amino acid for each codon, resulting in a protein with a specified sequence.
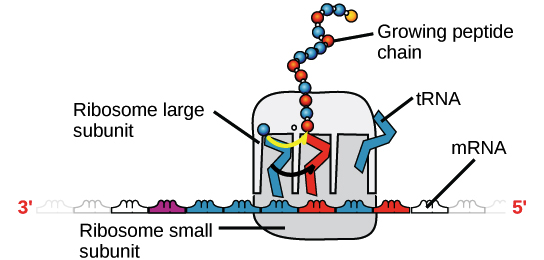
A ribosome (made up of proteins and rRNA) is coupled to an mRNA, with tRNAs delivering amino acids to the expanding chain. The sequence of the mRNA that is being "read" at the time determines the tRNA that binds and thus the amino acid that is added at that time.
Ribosomal RNA (rRNA) and transfer RNA (tRNA)
Ribosomal RNA (rRNA) is a key component of ribosomes, where it assists mRNA in binding to the correct location so that its sequence information can be read. Some rRNAs also function as enzymes, which means they aid in the speeding up (catalysing) of chemical reactions, such as the creation of bonds between amino acids to make proteins. Ribozymes are RNAs that function as enzymes.
Transfer RNAs (tRNAs) are also involved in protein synthesis, but their job is to act as carriers – to bring amino acids to the ribosome, ensuring that the amino acid added to the chain is the one specified by the mRNA. Transfer RNAs are made up of a single strand of RNA with complementary segments that connect to form double-stranded regions. This base-pairing results in a complicated three-dimensional structure that is crucial to the molecule's function.
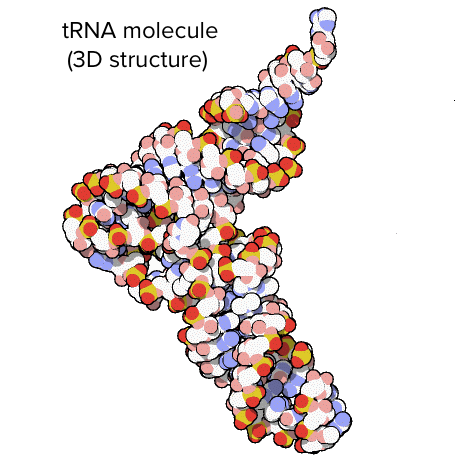
A tRNA's structure. The overall shape of the molecule is that of an L.
Regulatory RNA (miRNAs and siRNAs)
Non-coding RNAs (RNAs that do not code for proteins) play a role in regulating the expression of other genes. These RNAs are known as regulatory RNAs. MicroRNAs (miRNAs) and small interfering RNAs (siRNAs) are two examples. Small regulatory RNA molecules, or siRNAs, are around 22 nucleotides long. They attach to certain mRNA molecules (with partially or totally complementary sequences) and diminish their stability or interfere with translation, allowing the cell to reduce or fine-tune mRNA levels.
These are only a few of the many varieties of noncoding and regulatory RNAs that exist. Noncoding RNA continues to be discovered by scientists.
Key takeaway:
- DNA and RNA are the two main forms of nucleic acids, and both are made up of nucleotides, which have a five-carbon sugar backbone, a phosphate group, and a nitrogen base.
- RNA transforms DNA into proteins to carry out biological operations, while DNA provides the code for the cell's actions.
- An organism's characteristics are formed by the sequence of nitrogen bases (A, T, C, G) in DNA.
- In nucleic acid molecules, the nitrogen bases A and T (or U in RNA) always go together, and C and G always go together, establishing the 5′-3′ phosphodiester bond.
2.11.1 Nucleic Acids and component units:
Deoxyribonucleic acids (DNA):
Nucleic acids made up of nucleotide units having a 2-deoxy--D-ribosyl component. BNRD Rule N-1.3.
Nucleic acids: *
Macromolecules are the primary organic substance in biological cells' nucleus, made up of nucleotide units that can be hydrolyzed into pyrimidine or purine bases (typically adenine, cytosine, guanine, thymine, and uracil), D-ribose or 2-deoxy-D-ribose, and phosphoric acid. Nucleotides and ribonucleic acids are examples of nucleotides and ribonucleic acids, respectively. Rule N-1.3 of the BNRD (p. 110).
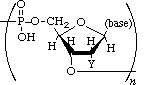 Y = H or OH
Y = H or OH
Nucleoproteins:
Proteins with nucleic acids as prosthetic groups, which when hydrolytically cleaved yield nucleic acids (or their cleavage products) as well as amino acids.
Nucleosides: *
Certain pyrimidine or purine bases' ribosyl or deoxyribosyl derivatives (occasionally, other glycosyl derivatives) The lack of phosphorylation makes them glycosylamines or N-glycosides, which are connected to nucleotides. Analogous molecules with the glycosyl group connected to carbon rather than nitrogen ('C-nucleosides') are now commonly included among nucleosides. Also see nucleic acids. N-2.3 of the BNRD (p. 110).
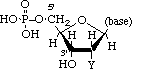
Y = H, a deoxyribonucleotide
Y = OH, a ribonucleotide
Nucleotides: *
Compounds formally obtained by esterification of the 3' or 5' hydroxy group of nucleosides with phosphoric acid. They are the monomers of nucleic acids and are formed from them by hydrolytic cleavage. BNRD Rules N-1.1, N-2.4
Y = H, a deoxyribonucleotide; Y = OH, a ribonucleotide
Oligonucleotides: * See oligo and nucleotides. BNRD Rule N-3.3
Purine bases:
Purine and its replacement derivatives, particularly those found in nature. The numbering in the table below is not in any particular order. B-2.11 of the NOC
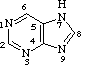
Purine
Pyrimidine bases:
Pyrimidine and its replacement derivatives, particularly those found in nature.
Pyrimidine
Ribonucleic acids (RNA):
Polyribonucleotides are naturally occurring polyribonucleotides. Nucleic acids, nucleosides, nucleotides, and ribonucleotides are all examples of nucleic acids. Rule N-1.3 of the BNRD (p. 110).
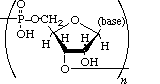
Ribonucleotides:*
Nucleotides with a ribosyl group as the glycosyl group. N-1.1 of the BNRD (p. 109). Also see nucleotides.
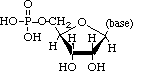
Key takeaway:
- The backbone of DNA is made up of sugar and phosphate, while the nitrogen bases are present in the centre and hold the two strands together, resembling a twisted stairway.
- The nitrogen bases can only be paired in specific ways: A with T and C with G. This is referred to as base pairing.
- The DNA strands are complementary to each other, run in opposing directions, and are referred to as antiparallel strands because of the base pairing.
2.12.1 Nucleic Acids- Nucleosides and Nucleotides
• A nucleotide is a monomeric unit of nucleic acids that serves as a source of chemical energy (ATP, GTP), participates in cellular signalling (cAMP, cGMP), and serves as an important cofactor in enzymatic reactions (coA, FAD, FM).
• A nucleoside is a molecule that lacks the phosphate group of a nucleotide.
• Nucleosides are simple glycosylamines made up of a nitrogenous base and a five-carbon sugar (either ribose or deoxyribose).
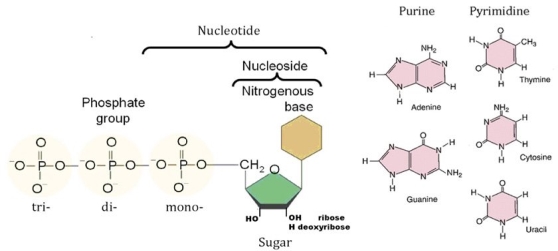
2.12.2 Structure of Nucleotides:
A nitrogen-containing base, a five-carbon sugar (pentose), and at least one phosphate group make up a single nucleotide. A nucleotide is also known as a "nucleoside phosphate" when all three are linked together.
Individual phosphate molecules bind the sugar-ring molecules in two adjacent nucleotide monomers repeatedly, forming a lengthy chain that connects the nucleotide monomers of a nucleic acid end-to-end.
Singular cyclic nucleotides are created when the phosphate group is bonded twice to the same sugar molecule, i.e., at the corners of the sugar hydroxyl groups, unlike nucleic acid nucleotides.
Nitrogenous bases
• In cells, the nitrogenous base is either a purine or a pyrimidine, and there are five main bases. Purine derivatives include adenine and guanine, while pyrimidine derivatives include thymine, cytosine, and uracil.
• Purines have two rings and include adenine and guanine.
• Adenine's rings have an ammonia group, whereas guanine's rings have a ketone group.
• Pyrimidines have one ring and contain cytosine, thiamine, and uracil.
• Thymine (found in DNA) and uracil (found in RNA) contain ketone groups in common, but thymine contains an additional methyl group on its ring.
• The three hydrogen bonds between guanine and cytosine are stronger than the two-hydrogen links between adenine and thymine (two hydrogen bonds).
Pentose Sugar
• Either a ribose (in RNA) or a deoxyribose (in DNA) molecule has five carbons.
• Both types of pentose sugars are found in nucleotides in their beta-furanose (closed five-membered ring) form.
2.12.3 Structure of Nucleosides
• A nucleoside has only a nitrogenous base and a five-carbon sugar, whereas a nucleotide has a nucleobase, a five-carbon sugar, and one or more phosphate groups.
• Nucleosides include cytidine, uridine, adenosine, guanosine, thymidine, and inosine, which are all connected to ribose or deoxyribose via a beta-glycosidic bond at the 1' position.
2.12.4 Properties of Nucleotides
Purine bases have certain properties.
• Light absorption in the UV area at 260 nm (detection and quantification of nucleotides) • Capable of generating hydrogen bonds
• Aromatic base atoms 1 through 9
• The purine ring is generated by the pyrimidine ring fusing with the imidazole ring.
• Numbering is done in a counter clockwise direction.
Adenine: Chemically it is 6-aminopurine
Guanine: Chemically it is 2-amino,6-oxy purine
Can be present as lactam & lactim form
Pyrimidine bases have certain properties.
• Capable of establishing hydrogen bonds
• Soluble at body pH
• Absorb UV light at 260 nm
• For pyrimidine, the aromatic base atoms are numbered 1 to 6.
• Base atoms have the same number of atoms or groups linked to them as the ring atom to which they are bound.
Cytosine: Chemically is 2-oxy ,4-amino pyrimidine
Exist both lactam or lactim form
Thymine: Chemically is 2,4 dioxy ,5-methyl pyrimidine
Occurs only in DNA
Uracil: Chemically is 2,4 dioxy pyrimidine
Found only in RNA
2.12.5 Properties of Pentose Sugars
• Ribose is the most prevalent pentose, with one oxygen atom connected to each carbon atom, and is a monosaccharide with five carbon atoms.
• Deoxyribose sugar is made by removing one oxygen atom from ribose sugar.
• Intramolecular hemiacetals are formed when the aldehyde functional group in carbohydrates reacts with neighbouring hydroxyl functional groups.
• The resulting ring structure is called a furanose because it is connected to furan.
• The ring opens and closes spontaneously, permitting rotation around the bond between the carbonyl group and the surrounding carbon atom, resulting in two different configurations. Mutarotation is the name given to this process.
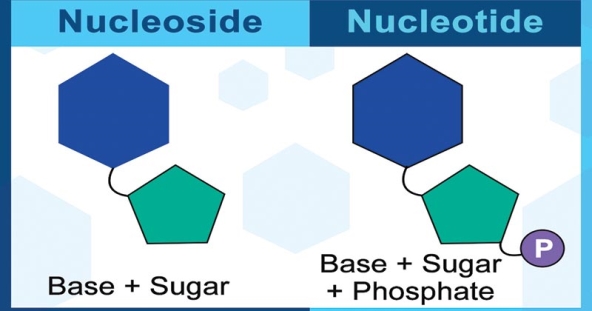
2.12.6 Classification of Nucleotides
On the basis of the type of sugar present, nucleotides may be:
- Ribonucleotides if the sugar is ribose.
- Deoxyribonucleotides if the sugar is deoxyribose.
2.12.7 Classification of Nucleosides
On the basis of type of nitrogenous bases present, nucleoside derivatives may be also grouped as following:
- Adenosine nucleotides: ATP, ADP, AMP, Cyclic AMP
- Guanosine nucleotides: GTP, GDP, GMP, Cyclic GMP
- Cytidine nucleotides: CTP, CDP, CMP and certain deoxy CDP derivatives of glucose, choline and ethanolamine
- Uridine nucleotides: UDP
- Miscellaneous: PAPS (active sulphate), SAM (active methionine), certain coenzymes like NAD+, FAD, FMN, Cobamide coenzyme, CoA
2.12.8 Functions of Nucleotides
• Because nucleotides are the building blocks of nucleic acids, the chemicals that control all hereditary features, they are extremely important to living creatures. Nucleosides are linked by 3′,5′-phosphodiester bridges to form polynucleotides. The genetic message is encoded in the base sequence of the polynucleotide chain. In cellular metabolism, nucleotides have a number of activities. In metabolic exchanges, they serve as the energy currency.
• They serve as crucial chemical linkages in the cell's response to hormones and other external stimuli.
• They're the building blocks of a variety of enzyme cofactors and metabolic intermediates.
• Every protein's structure, and eventually every biomolecule and cellular component's structure, is the result of information encoded in the nucleotide sequence of a cell's nucleic acids.
• Acting as energy storage for usage in phosphate transfer reactions in the future. ATP is primarily responsible for these reactions.
• Contributing to the formation of key coenzymes such NAD+, NADP+, FAD, and coenzyme A.
• Acting as second messengers in signal transduction events, as well as mediators of a variety of key cellular processes. Cyclic-AMP (cAMP), a cyclic derivative of AMP generated from ATP, is the most common second messenger.
• Acting as neurotransmitters and ligands for signal receptors. Adenosine is an inhibitory neurotransmitter, and ATP influences synaptic neurotransmission in both the central and peripheral nervous systems. ADP is a key stimulator of platelet activities, which helps to regulate blood coagulation.
• Using allosteric effects on enzyme activity to control a variety of enzymatic processes.
• Acting as activated intermediates in a variety of biochemical reactions. S-adenosylmethionine (S-AdoMet or SAM), which is engaged in methyl transfer processes, as well as the numerous sugar linked nucleotides involved in glycogen and glycoprotein production, are examples of activated intermediates.
2.12.9 Difference between Nucleotide and Nucleoside
Because they are both building elements in the creation process, it is critical to understand the key differences between nucleotides and nucleosides. The existence of Nucleic Acid and Genetic Code, which aids in the transfer of genetic information, is another vital factor that works as a building block.
Difference between Nucleotide and Nucleoside | |
Nucleotide | Nucleoside |
A phosphate group, a sugar, and a nitrogenous base make up a nucleotide's chemical makeup. | A sugar and a base lacking the phosphate group make up a nucleoside's chemical composition. |
They are still one of the leading causes of cancer-causing chemicals today. | They are largely employed in medicine to combat infections and cancer-causing substances. |
Adenosine, guanosine, and other nucleotides are examples of nucleotides. | Some of the most common nucleosides are nucleotides with phosphate groups added. |
Thus, these are the key differences between Nucleotides and Nucleosides.
Mechanisms of the Formation of Adenine, Guanine, and their Analogs in UV-Irradiated Mixed NH3: H2O Molecular Ices Containing Purine.
Introduction
A lot of scenarios for the emergence of life on Earth have been proposed. They entail prebiotic chemical reactions that eventually result in complex biomolecules capable of self-replication and evolution. Extra-terrestrial transfer of biogenic chemicals to early Earth has been proposed as a theory. If such extra-terrestrial transport is responsible for the synthesis of the earliest biomolecules and life on Earth, then analysing the molecular composition and evolution on the surfaces of meteorites and comets that frequently impact the planet can provide insight into such processes. A number of well-studied meteorites, such as Murchison, have been shown to contain biologically important compounds. Amino acids, carbohydrates, and nucleic acids, which are essential components of DNA and RNA as well as proteins, are among these compounds. This research focuses entirely on the purine-based nucleobases adenine and guanine, as well as their analogues, and their putative production processes.
Purine and pyrimidine are two forms of nitrogenated cyclic aromatic hydrocarbons or N-heterocycles that can be used to make nucleic acids. The presence of nitrogenated heterocycles in the gaseous interstellar medium, in thick and diffuse clouds, has long been suspected, but never verified. Despite exhaustive searches, pyrimidine (C4H4N2), a nitrogenated monocyclic molecule, has not been found in the interstellar medium in the gas phase. However, pyrimidine cation production via ion-molecule reactions has been observed in the gas phase. Purine, a pentamer of HCN, is formed by combining five hydrogen cyanide molecules and is projected to be exceedingly exothermic, hence it is unlikely to occur in isolation.
Meteorites, on the other hand, have been shown to contain nitrogenated cyclic compounds. Adenine, guanine, cytosine, and uracil were recovered from the well-known Murchison and Murray carbonaceous chondrites and shown to be of extra-terrestrial origin. The meteoritic samples also included nucleic acids, amino acids, and other chemical substances of biological relevance. It is unknown whether they are synthesised in the gas phase and supplied to the grain surface, or whether they are formed on the cold grain surfaces as a result of photo-processing. However, once distributed on grain surfaces, these biomolecule precursors have been proven to be photo-processed into larger, more complicated molecules. The photo-processing of ices deposited on grain surfaces by cosmic radiation is a probable method for their development. These ices are known to contain volatile organic molecules such as H2O, CH3OH, CO, CO2, NH3, CH4, as well as larger molecules such as PAHs and nitrogenated heterocycles.
Materese, Nuevo, and Sandford conducted a laboratory study on purine photo-processing in astrophysical ices. They used a substrate chilled to 15K to deposit a combination of H2O, NH3, and purine (relative abundances of roughly 1:0.1:10-3). Simulating astrophysical photo-processing, the expanding ice was bombarded with UV light (Lyman at 121.6 nm, i.e., 10.2 eV, and a broad-band continuum centred at 160nm). Gas-chromatographic (GC-MS) techniques were used to examine materials after 1–2 days of irradiation. Materese, Nuevo, and Sandford (Materese et al. 2016) discuss their experimental findings in a companion publication. Materese et al. Discovered that 2-aminopurine, 6-aminopurine (adenine), 2,6-diminopurine, hypoxanthine, xanthine, isoguanine, and guanine are all produced in measurable amounts. It had previously been discovered that irradiating pyrimidine in analogously formed ices created uracil, cytosine, and thymine (albeit only minute amounts of the latter after high doses of radiation), and the mechanics of their synthesis had been explored.
We investigate the methods by which doped purine could be aminated and hydroxylated in the irradiation NH3: H2O experimental ices using electronic structure simulations. We look at the physical processes that could occur under the experimental settings, as well as the intermediates that could be produced and the reactions that they could experience. Independent of experimental input, the results form reaction pathways leading from irradiation reactants to a variety of products. The computational results are then compared to the experimental results. With the studies identifying many (though not all) major products of purine photo-processing in astrophysical ices, our computational findings may aid in deciphering the preferred reaction routes and the preference for certain products over others. In order to obtain insight into the nature of photochemical transformations that occur on cold grain surfaces or tiny solar system objects such as asteroids and comets, we explore the roles played by physical factors such as UV irradiation and condensed phase environment in this transformation.
Methods
Density functional theory (DFT), the most extensively used class of electronic structure approaches, was utilised to calculate the structures and relative energies of the reactants, intermediates, and products. Geometry optimizations were done specifically with the B3LYP functional and the cc-pVDZ basis set. Using B97M–V, a range-separated hybrid meta-GGA functional with the cc-pVQZ basis set, higher accuracy relative energies of the structures were estimated. Across the main group elements, B97M–V has been proven to be quite accurate for non-covalent interactions, thermochemistry, barrier heights, and more. It also lowers self-interaction error as a range-separated functional, making it ideal for the description of radical cations.
We employed the conductor-like solvent model C-PCM from the class of polarizable continuum models to model the influence of the extended ice environment (PCM). In particular, we used an implementation that assures constant potential energy surfaces while molecular shape changes. The bulk is represented in this model as a polarizable medium with a dielectric constant of. The solvent dielectric should better simulate the condensed phase, allowing us to investigate the effect of the solvent. The geometries were re-optimized with the solvent model in the PCM calculations, again using B3LYP/cc-pVDZ. Single point PCM energies were then calculated at the ωB97M–V/cc-PVQZ level of theory. We used ε= 78.39 for the dielectric constant of water for all calculations.
All calculations used the Q-Chem 4 quantum chemistry program package.
Results
Organic molecules undergo a variety of physical and chemical changes when exposed to UV light directly. Purine will undergo electronic excitation into excited states, simultaneous excitation of vibrational and rotational modes, and ionisation to produce cations and free electrons when exposed to UV light. Isomerization and photo-fragmentation may occur as a result of excess energy in excited states and/or ions. Irradiation of the ices can yield a variety of reactive species, including OH and NH2 in H2O and NH3 ices, respectively, or both in a mixed molecular ice. It can also produce electrically excited solvent molecules that breakdown into fragments or release energy through collisions and radiative energy release.
In the UV-wavelength region of the H2 UV source used in the experiments, H2O has a significant photo-dissociation cross-section (Materese et al. 2016). Around 181 nm, NH3 dissociates into NH2 and H, with a non-negligible dissociation cross-section. Additionally, hydroxyl radicals can remove hydrogen from NH3 and form NH2 radicals. In summary, the following processes have the ability to produce hydroxyl (OH) and amino (NH2) radicals in ice:
(1) H2O+hν→H∙+O∙H
(2) NH3+hν→H∙+N∙H2
(3) H2O + hν → H2O•+ + e-
(4) NH3 + hν → NH3•+ + e-
(5) NH3+O∙H→N∙H2+H2O
Of the above-mentioned photo-processes all channels but (3) seem open under Lyman α irradiation. Purine can also produce radicals and ions through the following processes:
(6) Pu + hν → [Pu]+• + e-
(7) Pu + hν → [Pu/H]• + H•
Where, [Pu/H]• designates a radical formed by the cleavage of a C–H bond on the purine ring.
The main question is how hydroxyl and amino modified purine derivatives are created from the parent neutral molecules and the radicals and ions produced in the primary photoinduced reactions mentioned above. Furthermore, ionization energy of NH3 is 10.02 eV, which is low enough for Lyman-alpha (121.6 eV, ~10.2 eV) to ionize it to NH3+ have previously shown that hydroxyl radical abstracts hydrogen atoms from NH3 to produce NH2 easily (reaction 5 in the manuscript). As H2O is ten times more abundant than NH3 in the experimental conditions, hydroxyl radicals would be produced in large quantities, which would produce NH2 radicals. Direct dissociation would be used to supplement the basic NH2 production process. As a result, we expect that NH2 will be the most prominent nitrogenous attacking group, albeit interactions involving NH3+ are not impossible. The progressive substitution of NH2 and OH groups on the purine ring is summarised in Fig. Let's start by listing some of the most common reaction pathways. To begin with, neutral purine can react with neutral H2O or NH3, as well as the OH and NH2 radicals. Purine cations can also react with H2O or NH3, as well as the OH and NH2 radicals. Third, purine radicals (those without a H atom) can recombine with OH or NH2 radicals. Other options are also available.
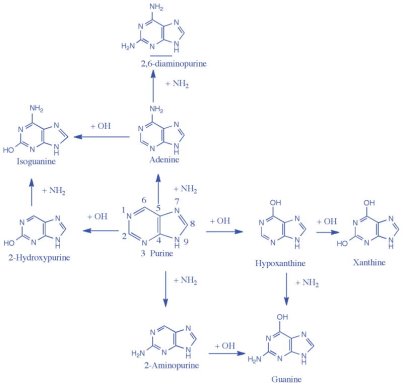
The amino and hydroxyl group substitutions on the purine ring are shown in this chemical scheme. Vertical arrows represent amino group alterations, whereas horizontal arrows represent hydroxyl group substitutions. The most advantageous singly substituted photo-products are 2-hydroxypurine and adenine, while the most advantageous doubly substituted photo-products are xanthine, isoguanine, guanine, and 2,6-diaminopurine.
(8) Pu + H2O → [Pu/H - OH] + H2
(9) Pu + NH3 → [Pu/H - NH2] + H2
(10) Pu+O∙H→[Pu/H−OH]+H∙
(11) Pu+N∙H2→[Pu/H−NH2]+H∙
(12) Pu•+ + H2O → [Pu/H - OH] + H2+
(13) Pu•+ + NH3 → [Pu/H - NH2] + H2+
(14) Pu∙++O∙H→[Pu/H−OH]+H+
(15) Pu∙++N∙H2→[Pu/H−NH2]+H+
(16) Pu/H∙+O∙H→[Pu/H−OH]
(17) Pu/H∙+N∙H2→[Pu/H−NH2]
Because of their large activation barriers, neutral–neutral reactions (8 and 9) between H2O (or NH3) and purine are projected to be several orders of magnitude slower than reactions involving radicals or cations, especially in cold ice matrices. As a result, it's plausible to predict that the interactions of OH and NH2 radicals with ionised, radical, or neutral purine, and its derivatives, will dominate the chemistry in these ices.
We looked at the neutral purine and hydroxyl radical (and amine radical) routes (10 and 11) and discovered that adding the hydroxyl (or amino) radical to purine, which results in radical intermediates, is endothermic because aromaticity is lost. As a result of this finding, we believe channels 10 and 11 are less likely than some of the other alternatives outlined below. After the loss of a H atom, the reactions are exothermic, yielding neutral substituted products.
Because the UV-lamp has enough energy to ionise purine, purine cations are likely to occur in the irradiated mixed molecular ices (primary channel 6). With nucleophilic H2O or NH3, the purine cation can undergo an addition process (secondary channels 12 and 13). Because this reaction is both exothermic and barrierless in the entrance channel, it is likely to proceed. As an example, consider the following reaction with the water molecule:
(18) Pu•+ + H2O → [Pu-OH2]•+.
Furthermore, because only one principal photoproduct is required for this reaction to continue, it is likely to occur under low photon flux. To make the final hydroxylated or aminated products from the early intermediates, the process requires the removal of a proton and an H-atom (or maybe H2+•). As a result, as seen in Fig, channels 12 and 13 have several steps. The loss of the H atom from the intermediates is one of these stages that has a high reaction barrier and hence is unlikely to occur directly. If the H atom is later taken by another radical, R (H or OH or NH2), the barrier is decreased and possibly abolished entirely:
(19) [Pu-OH2]•+ + R• → [Pu-OH]+ + RH.
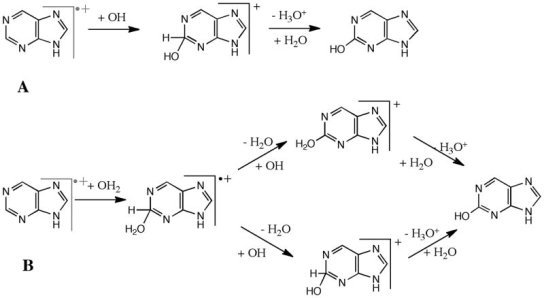
The addition of OH (A) and H2O (B) to the purine radical cation has two mechanisms. The addition of hydroxyl with purine cation is the initial step in A, followed by the loss of a proton aided by the H2O molecule (s). The addition of H2O with purine radical cation is the initial step in B, followed by the sequential loss of a hydrogen atom and proton.
Finally, in a third reaction, proton abstraction would provide the end product:
(20) [Pu-OH]+ + H2O → [Pu/H - OH] + H3O+.
Pathway A, shown in Figure, is a nucleophilic substitution pathway that involves secondary reactions 14 and 15 between the purine radical cation and the OH radical (or NH2 radical). When an electron from purine's -electron system is ionised, it leaves a hole in the aromatic system. The charge is most likely concentrated on the carbon atoms that are less electronegative. The two carbon atoms (C2 or C6) on the six-membered aromatic ring, or the C8 carbon atom on the five-membered ring, can then be attacked by a radical NH2 or OH group. Theoretically, addition to the nitrogen atoms on the ring is also possible. Because it requires the simultaneous existence of two reactive intermediates obtained from the principal photochemical routes, this channel is most likely to proceed under high radiation circumstances. In the entrance channel, it was previously demonstrated that a reaction between a polycyclic aromatic hydrocarbon and hydroxyl is exothermic and barrierless.
The intermediates associated with reactions 18–20 is depicted in Figure of route B. After adding H2O, the radical cation complex 18, loses a hydrogen atom and a proton in a gradual manner (concentrated H2+ loss is uncommon) to generate hydroxypurine (Figure B). This can happen as a result of H abstraction (19), followed by H+ loss (20), or it can happen in the opposite sequence. It's worth noting that H (or H+) can be extracted from two sources: CH at the sp3 site, or H2O, as shown in Fig. According to our calculations, H is abstracted first using an OH radical to generate a closed-shell cationic intermediate. Similar to pathway A, this intermediate undergoes proton loss with the help of the surrounding H2O molecules.
We previously discovered that the ionic pathway was the most critical and efficient when exploring similar mechanisms employing pyrimidine (C4H2N2) molecules. Ionizing radiation causes nucleobases to undergo a variety of bond-breaking and structural changes. The results of the cationic pathways will be discussed in this study from now on (coming from 14 and 15 directly, or from 12 and 13 indirectly). In section IV, we go through why the ionic mechanism is more likely than other sorts of mechanisms in mixed molecular ices.
Dehydrogenated purine radicals (Pu/H) can react with OH or NH2 radicals to form hydroxylated or aminated products when CH or NH bonds in purine are broken (16 and 17). We discovered that these reactions are exothermic and can directly manufacture the products. However, because they necessitate the presence of two reactive species created by primary radiative processes at the same time, they will be useful only when the concentration of such species approaches that of the parent species. Under strong radiation exposure, H atoms formed from H2O and NH3 can, of course, combine with Pu/H to regenerate neutral purine. The hydrogen atoms can also combine with purine neutral to form hydrogenated purine.
The OH radical has the potential to further oxidise purine oxidation products, resulting in doubly oxidised compounds. This could happen after (as in channel 21) or before (as in channel 22) ionisation of the monohydroxy product:
(21) [Pu/H−OH]+∙+O∙H→[Pu/H/H(−OH)2]+H+
(22) Pu/H−OH+O∙H→[Pu/H(−OH)2]∙→[Pu/H/H(−OH)2]+H∙.
In the entrance channel, the above ion-radical and radical-molecule processes are typically rapid and barrierless. Proton loss from protonated intermediates in aqueous media is spontaneous, as previously demonstrated.
We looked into these pathways to see if the interactions between these reactants that lead to functionalized products are beneficial, and if adenine and guanine creation is preferred over other possible products. The catalytic activity of H2O, NH3, and mixed molecular ices was also investigated. The laboratory experiment findings were interpreted using this quantum chemical data.
The stepwise substitution of NH2 and OH groups on the purine ring, which leads to major singly and doubly substituted products, is summarised in Fig. The hydroxyl (OH) group substitution on the ring is indicated by the horizontal arrows, whereas the amino (NH2) group substitution is indicated by the vertical arrows. Oxidized purines such as hypoxanthine (6-hydroxypurine) and 2-hydroxypurine (also known as purinol) are formed after the initial OH substitution on the six-membered ring. Xanthine would result from an extra hydroxyl substitution at the C2 site (2,6, -dihydroxypurine). In the experimental samples produced by UV-irradiation of H2O: purine and H2O: NH3: purine mixed molecular ices, both hypoxanthine and xanthine were found. Guanine is formed when an NH2 group is substituted for hypoxanthine, while isoguanine is formed when an NH2 group is substituted for 2-hydroxypurine. 2-aminopurine and adenine (6-aminopurine) are produced by adding an NH2 group to the 2, or 6 places first on the purine ring, respectively. 2,6-diminopurine is formed by adding a second NH2 group to adenine on the six-membered purine ring. Isoguanine is made by adding a hydroxyl group to adenine, and guanine is made by adding a hydroxyl group to 2-aminopurine. After irradiating purine in mixed molecular ices, 2,6-diaminopurine, guanine, and isoguanine were all found in the experimental samples. The next sections take a step-by-step look at these reactions using the reaction mechanisms outlined above.
Step 1: 1st Substitution
Amine group substitution to purine
Figure shows how an NH2 group can bind barrierless to the purine cation's carbon structure to produce three intermediates. The reactants are 57.8 and 54.0 kcal/mol below the 2(2H)-aminopurine and 6(6H)-aminopurine cations (labelled 3b and 3a in Fig). The lowest energy intermediate is the 8(8H)-aminopurine cation (labelled 3c in Fig), which is 68.2 kcal/mol lower than the reactants, which are a bare purine cation, H2O, and an NH2 radical. In other words, the 8(8H)-aminopurine molecule has a higher proton affinity than the 2- and 6-aminopurine molecules because the aromaticity of the six-membered ring is preserved when the proton attaches to the carbon framework. To make 2-, 6-, and 8-aminopurine, a proton will be lost. The energy of the products is higher than the isolated reactants devoid of H2O, hence a purely gas phase proton loss is energetically unfavourable. The combined energy of the products and an H3O+ falls below that of the reactants and intermediates when an H2O molecule is explicitly included and the proton is permitted to be stolen by an H2O molecule, as shown in Fig. This is comparable to the pyrimidine oxidation reaction, which we discovered needs the presence of numerous H2O molecules capable of removing the proton from the charged intermediates. The lowest energy product, adenine, 6-aminopurine, is around 73.1 kcal/mol lower than the reactants. 2-aminopurine is slightly more energetic than adenine, while 8-aminopurine is even more energetic. After the first amino group is added to purine, the most likely amine result is adenine, which can only be produced in the presence of a mixed H2O-NH3 ice.
On the left, a chemical diagram for the cationic mechanism of amino group substitution to purine, starting with purine cation, NH2, and H2O as reactants, with zero kcal/mol arbitrarily set. The structures in square brackets are intermediates, and the structures on the right are products. The black values were computed in the gas phase using wB97M-V/aug-cc-pVQZ, whereas the red values were derived in the condensed phase using a polarizable continuum model.
Allowing this reaction to take place in a condensed phase environment offers new information about the process. A modified polarised continuum model with a dielectric to mimic the bulk water was used to treat the reactants, purine cation, hydroxyl radical, and one molecule of H2O. The neutral products plus the hydronium ions were treated in the same way as the charged closed shell intermediates, and their energies are shown in Fig. In the presence of the solvent, the red numbers were determined for the reactants, intermediates, and products. The energy of the reactants, intermediates, and products are all reduced by the presence of the solvent, but not in the same way. In Fig, the total energy of the reactants (purine cation, H2O, and NH2 radical) was selected as zero. The cationic intermediates 2-, 6-, and 8-aminopurine cations, respectively, are 54.0, 50.7, and 62.8 kcal/mol lower than the reactants. In the solvent, they are less stabilised in relation to the reactants than in the gas phase. This suggests that the solvent stabilises the reactants slightly better than the intermediates. The final products 2-, 6-, and 8-aminopurines have kcal/mol values that are 97.3, 99.7, and 95.9 kcal/mol lower than the reactants, indicating that the products are substantially more solvent-stabilized than the intermediates. Due to solvation, the relative energy of the products is reduced by about 30 kcal/mol. In this scenario, gas phase simulations show that only one H2O molecule is required to make the reaction favourable. The solvent phase calculations confirm the gas phase observations - that the forward reaction is possible in the presence of H2O, as seen in Fig.
Finally, we remark that different intermediates are certainly feasible, which is in line with the preceding consideration of various mechanistic routes. At the C2, C6, or C8 locations, ammonia can form a compound with purine cation. H can be abstracted from the sp3 CH bond or from NH3 by another radical (e.g., OH). Due to the basicity of amines, the latter procedure produces intermediates 3a, 3b, and 3c, whereas the former method produces lower energy intermediates. Proton migration from 3a, 3b, and 3c may also make these protonated aminopurines accessible.
Hydroxyl group addition to Purine
Of course, the hydroxylation process competes with the amination in the first step in an H2O: NH3: purine mixed molecular ice. In recent experiments, the H2O: NH3 ratio was found to be around 10:1. (Materese et al. 2016) As a result, the OH radicals formed by H2O are more likely to oxidise purine first. Following a cationic mechanism, we investigated the energies of the hydroxylation process. As shown in Fig, OH attack on the purine cation can occur at three different carbon atoms, yielding 2(2H), 6(6H), and 8(8H)-hydroxypurine ion intermediates (labelled as 4b, 4a and 4c respectively). To produce the products, these intermediates lose a proton. The lowest energy intermediate is the 8-hydroxypurine cation, which is 69.3 kcal/mol less than the reactants, followed by the 2-hydroxypurine cation and the 6-hydroxypurine cations, which are 60.9 and 53.9 kcal/mol less than the reactants, respectively. The hydronium ion (H3O+) and three products are produced when an H2O molecule expressly abstracts the proton from the intermediates. 2-hydroxypurine, 6-hydroxypurine, and 8-hydroxypurine have combined energies of 77.2, 76.2, and 74.4 kcal/mol, respectively. If one follows a pure gas phase mechanism, the 2-hydroxypurine is the most advantageous isomer among the three mono-hydroxylated products investigated in Fig.
On the left, a chemical diagram for the cationic mechanism of hydroxyl group substitution to purine, starting with purine cation, OH, and H2O as reactants, with zero kcal/mol arbitrarily set. The structures in square brackets are intermediates, and the structures on the right are products. The black values were computed in the gas phase using wB97M-V/aug-cc-pVQZ, whereas the red values were derived in the condensed phase using a polarizable continuum model.
The hydroxylation of pyrimidine molecules differs in that the hydroxy-purine cationic intermediates are significantly easier to deprotonate. Multiple H2O molecules, i.e., a condensed phase-like environment, were required for pyrimidine hydroxylation to happen. In the instance of purine, we can see that only one H2O molecule is required to complete the process. However, the reaction nevertheless proceeds more quickly in the ice matrix than in the gas phase via a solely bimolecular process, as demonstrated below.
The intermediates 8(8H)-hydroxypurine, 2(2H)-hydroxypurine, and 6(6H)-hydroxypurine cations (4c,4b, and 4a correspondingly in Fig) have energy that are 65.7, 59.7, and 55.2 kcal/mol lower than the reactants in the condensed phase. The three products, 2-hydroxypurine, 6-hydroxypurine, and 8-hydroxypurine, as well as H3O+, have kcal/mol values of 104.1, 103.4, and 100.3, respectively, below the reactants. In the condensed phase, the products are stabilised by roughly 30 kcal/mol, owing to charge delocalization in the water matrix. The charge on the purine cation is more confined in the gas phase. The H2O matrix helps extract the proton from the intermediate and facilitates the process, according to the condensed phase calculations. In both gas phase and condensed phase computations, the relative energy ordering of the three products remains the same.
After the first stage, we'll look at adding amine and hydroxyl groups to the desired compounds in the second step.
Step 2: 2nd substitution
Following the addition of amine and hydroxyl groups to the purine ring, the process may proceed with more amine or hydroxyl group substitutions. The addition of NH2 to 2, and 6-hydroxypurine, as well as OH to 2, and 6-aminopurine, is discussed in the next section. The addition of an amine group to mono-aminopurine and a hydroxyl group to hydroxy-purine is then considered.
Amino group addition to 2, and 6-hydroxypurine(s)
In Fig, we look at the reactions of the 6-hydroxypurine and 2-hydroxypurine cations with the NH2 radical, using mechanisms identical to those we looked at in the previous steps. The total energy of the 6-hydroxypurine radical cation, the NH2 radical, and H2O in Figure is set to zero. The energies of all other species are displayed in relation to these zero energies. The 2-hydroxypurine cation has a lower energy than the 6-hydroxypurine cation by 6.3 kcal/mol. In Fig, the energies and structures of the intermediates and products derived from the 2-hydroxypurine cation are shown in blue. The cationic intermediates formed after the NH2 radical nucleophilically attacks the 6-hydroxypurine cation have 46.9 and 65.8 kcal/mol less than the reactants (colored black in Fig). The two products, guanine and 8-amino-6-hydroxypurine, are 63.9 and 58.3 kcal/mol lower than the reactants after proton loss to a neighbouring water molecule. Guanine (along with H3O+) is below the reactants and the proton-attached intermediate (5a) in terms of energy; the other intermediate (5b) is below the deprotonated product by 7.5 kcal/mol.
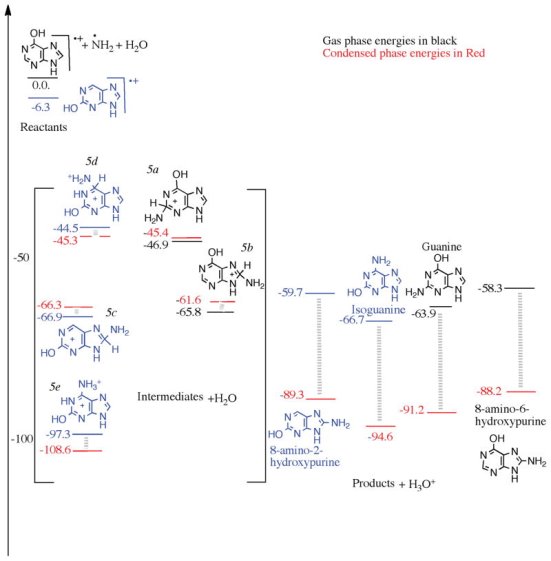
On the left, a reaction diagram for the cationic mechanism of amino group substitution to 2-hydroxypurine and 6-hydroxypurine starting with respective cations, NH2 and H2O as reactants, with zero kcal/mol arbitrarily set. The structures in square brackets are intermediates, and the structures on the right are products. The black numbers were computed in the gas phase using the wB97M-V/aug-cc-pVQZ level of theory, whereas the red numbers were derived using a polarizable continuum model in a condensed phase environment.
Structures 5c (66.9 kcal/mol), 5d (44.5 kcal/mol), and a rearranged version of 5d at 97.3 kcal/mol below the reactants are some of the intermediates associated with NH2 attack on the 2-hydroxypurine cation. The second intermediate, isoguanine protonated at the amine group (5e), has an extremely low energy compared to the other intermediates because the proton has migrated from the carbon atom (C6) to the amine group, giving the nitrogen atom a formal positive charge. This is also the structure that occurs when NH3 attacks the C6 site of the 2-hydroxypurine cation directly. Similar rearrangements of intermediates 5a, 5b, and 5c are feasible, and should be much lower in energy due to the relatively high basicity of amines. When a proton is lost, the lowest energy intermediate generates isoguanine, which has a relative energy of 66.7 kcal/mol lower than reactants, and the other intermediate generates 8-amino-2-hydroxyl-purine, which has a relative energy of 59.7 kcal/mol lower than reactants. On the six-membered ring, mono-hydroxylated purines prefer to attach an amine group. After amine group addition, 2-hydroxypurine creates isoguanine and 6-hydroxypurine creates guanine, which is the preferred result during the first hydroxylation step.
The combined energy of the reactants, namely the 6-hydroxypurine cation (or 2-hydroxypurine cation), the OH radical, and one H2O molecule determined in the presence of the solvent, have been reset to zero in the condensed phase. Figure also shows the relative energy of the intermediates (OH group attached cationic intermediates and H2O) and products (mono-amino mono hydroxy purine and H3O+). One of the intermediates (5e) in the gas phase is abnormally stable due to the proton's transfer from carbon to the NH2 group. With the exception of one situation in which the proton has relocated, the products have lower energy than the reactants and intermediates. The loss of a proton from the C6 carbon, on the other hand, is energetically plausible and likely, because to isoguanine's high gas phase proton affinity.
Hydroxyl group addition to 2, and 6-aminopurine(s)
A mono-hydroxylation yields four cationic intermediates from 2-aminopurine and 6-aminopurine cations. The two intermediates formed from the 6-aminopurine and hydroxyl radical, 2(2H)-hydroxy-6-aminopurine and 8(8H)-hydroxyl-6-aminopurine are 41.1 and 65.3 kcal/mol below reactants (colored black). The intermediates 6c and 6d (coloured blue) formed by the 2-aminopurine cation and the OH radical have 71.2 and 87.1 kcal/mol less than the reactants. As shown in Fig, these energies are lower than the product energies. Guanine, which is 51.5 kcal/mol below the reactants, and 8-hydroxy-2-aminopurine, which is 41.5 kcal/mol below the reactants, are the two products formed from the mono-hydroxy-2-aminopurine intermediates. Isoguanine, which is 54.3 kcal/mol below the reactants, and 8-hydroxy-6-aminopurine, which is 43.5 kcal/mol below the reactants, are the products associated with the 6-aminopurine. When only one water molecule is present, the products are not below the intermediates, while being substantially below the reactants. To deprotonate the intermediates and create the products, a water environment is required. Mono aminated purines, once again, prefer to attach a hydroxyl group to the six-membered ring, particularly on the 2 or 6 carbons. Isoguanine has a modest energy advantage over guanine. The experimental samples contain both isoguanine and guanine, with isoguanine being more prevalent than guanine.
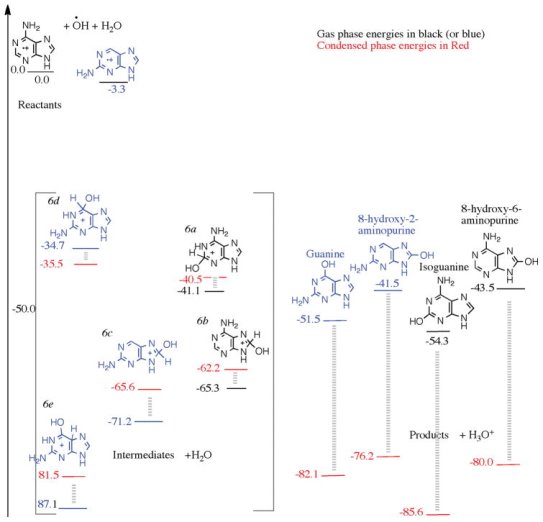
On the left, a reaction diagram for the cationic mechanism of amino group substitution to 2-aminopurine and 6-aminopurine starting with respective cations, OH and H2O as reactants, with zero kcal/mol arbitrarily set. The structures in square brackets are intermediates, and the structures on the right are products. In the gas phase, the black numbers were computed using the wB97M-V/aug-cc-pVQZ level of theory, whereas the red numbers were derived using a polarizable continuum model in a condensed phase environment.
When performing condensed phase calculations on the process, we start with the reactants 2-aminopurine and 6-aminopurine cations, OH radical, and H2O. Along with H3O+, mono-hydroxy-mono-aminopurines are produced. The H2O solvent effect stabilises the products by roughly 30 kcal/mol when compared to the gas phase. The intermediates are roughly the same as they were in the gas phase. In the condensed phase, three of the four products had energies lower than the intermediates depicted in Figure.
One intermediate (produced from 6d by proton migration from C6 to C5, but not depicted in Fig. Has a higher degree of stabilisation and is 83.5 kcal/mol lower than the reactants. This is because the structure is allowed to pucker slightly in the gas phase. The energy of this intermediate (plus an H2O) is comparable to that of the products guanine and H3O+. Other lower-energy protonated intermediates may be attainable if solvent assisted proton migration from the hydroxyl attack site to a more basic amine site is conceivable. This produces amine-protonated guanine or isoguanine, among other things. Protons could be shuttled more easily in an ice environment. Finally, we discover that isoguanine has a lower energy than guanine, both in the gas phase and in the condensed phase calculations. The products' relative energy ordering is also unchanged.
Amine group non-addition in pure ammonia ice
Except for a minute amount of adenine, no products were identified when the experiments were carried out in pure ammonia ice. The pyrimidine irradiation of pure ammonia ices yielded comparable effects. In contrast to the numbers of products formed in the mixed H2O and NH3 molecular ices and pure H2O ices, this absence of product formation is striking. The UV-source utilised for the irradiation (10.2 eV) is powerful enough to ionise NH3 to its cation (gas phase threshold is 10.1 eV) and photolyze it to create NH2 radicals (NH2), as previously discussed. A reaction mechanism might be proposed in which a purine radical cation combines with an amine (NH2) radical to form early intermediates, which then lose a proton to nearby NH3 molecules to form products and the NH4+ ion. Because NH3 has a high proton affinity of 211 kcal/mol and there are 10 NH3 molecules for every purine molecule in pure NH3 ices, this reaction sequence is plausible. However, the absence of experimentally identified products in pure ammonia ice shows that this pathway is blocked by some aspect of the ammonia ice.
Cuylle et al. Found that in Lyman irradiated ammonia ice seeded with pyrene, its anions are exclusively created, as a result of free electrons generated by ionisation of NH3, and the fact that the ratio of NH3 to pyrene was considerable, in a comparable ice experiment (5000:1). Pyrene cations are formed in water ices (also 5000:1), apparently by direct ionisation because Lyman does not ionise water molecules, and hence stray free electrons are not available. The outcomes of mixed H2O and NH3 ices with a 5000:1 ice: pyrene ratio was dependent on the respective abundances of H2O and NH3. The anion signal was quenched and only pyrene cations were created under conditions similar to the experiments we're replicating (H2O: NH3 ratio of 10:1).
Purine has a higher electron affinity than pyrene, hence in Lyman's irradiated NH3 ice tests, purine anions would be the only ones produced. From an electrostatic standpoint, a reaction of the purine anion with NH2, or other radicals, or the creation of a coordination complex with NH3, would be unfavourable. This is most likely why no product production was found in purine in NH3 ice in experimental tests. (Materese et al. 2016, Matarese et al., Matarese et al., Mat We expect purine cations to be present in a mixed ice with a 10:1 H2O: NH3 ratio, and the cationic reaction process to give all of the products predicted in this study or observed in the experiments.
Discussion
Surprisingly, the amine group likes to connect at position 6 on the purine ring, whereas the hydroxyl group prefers to bind at position 2. As a result, adenine is the favoured mono-aminated product thermodynamically, while 2-hydroxypurine is the favoured mono-hydroxylated product. After the first step, the upwards or left routes appear to represent the way ahead in Fig.
Both the NH2 and the OH radicals are powerful nucleophiles. According to our estimates, the reaction energy of purine with either NH2 or OH do not change significantly. After the initial round of substitutions, both mono-aminated and mono-hydroxylated products are expected to occur. The first NH2 substitution results in adenine, while the first OH substitution results in hypoxanthine, according to thermodynamics. As a result, adenine and 2-hydroxypurine are expected to be the most favoured products after the first step, while 2-aminopurine and 6-hydroxypurine, i.e., hypoxanthine, are also expected to form because their energies are relatively near and there is no barrier to these reactions. Because H2O is 10 times more plentiful in the reaction mixture, hydroxylation has a better likelihood of occurring, hypxanthine and 2-hydroxypurine are more likely to form. Hypoxanthine was discovered to be the most prevalent product in the laboratory, followed by adenine; 2-hydroxypurine could not be analysed since a standard was unavailable. Nonetheless, the most popular items provide a roadmap for future reactions.
If the NH2 group is preferred over the OH group, then after the initial NH2 group addition, adenine would be the energetically most-favored product, and it would be a sensible location to start for future NH2 or OH group addition. Isoguanine, which is one of the primary products, is formed when an OH group connects to adenine at the 2-position. If we start with 2-aminopurine, the most preferred product following the OH group is guanine. In condensed phase calculations, isoguanine has a lower energy than guanine by 3.5 kcal/mol, and hence should be an energetically favoured product. Of course, it can generate 2,6-diaminopurine by adding another NH2 group, and it does so in the trials. As a result, while there is no direct method to making guanine from adenine, there is one to generate isoguanine, which is why isoguanine is a prominent product in the experiment.
If, in the first phase (following the horizontal lines in Fig), OH group addition is preferred over NH2 group addition, we can expect to find either 2-OH-purine or hypoxanthine. The most popular product, 2-hydroxypurine, can be used as a starting point for further functionalization. As seen in Fig, adding an NH2 group to 2-hydroxypurine produces isoguanine rather than the other products. We acquire guanine if we start with hypoxanthine, which is a less preferred result after the first hydroxylation.
As a result, starting with either adenine or 2-hydroxypurine as the principal results of the first step, we can see that isoguanine is the preferred product. After the first stage, we must begin adding the OH or NH2 group to the less preferred products in order to make guanine. Because of the difficulties in forming guanine, this predicts that isoguanine would be substantially more abundant than guanine. Materese et al. (Materese et al. 2016) discovered that the singly substituted compounds hypoxanthine, adenine, and 2-aminopurine are the most prevalent products in an experimental setting. Products that are doubly substituted are order(s) of magnitude less numerous than those that are singly substituted. In order of abundance, xanthine, isoguanine, guanine, and 2,6-diaminopurine are the most abundant doubly substituted products.
Validation of cationic mechanisms for hydroxylation and amination
The absence of products in pure NH3 samples supports the cationic reaction mechanism over other types of reactions. We would expect to observe products generated in irradiated pure NH3 ices if other mechanisms were responsible for hydroxylation and amino group substitution, or if they were dominant over the cationic pathway. We can conclude that anionic purine does not lead to hydroxyl or amine substitution, at least not in an efficient manner, because products are not generated in pure NH3 ices. Positive discovery of products in 10:1 H2O: NH3 and pure H2O irradiated ices, where purine cations would be formed, as demonstrated by Cuyille et al., strongly suggests that the reaction proceeds via the cationic mechanism. Ions are also thought to have a role in the addition of hydroxyl groups to polycyclic aromatic hydrocarbons.
Astrobiology Significance
Our findings suggest that under UV irradiation, the nucleobases adenine and guanine can be photochemically produced in the condensed phase in a mixture of purine, water, and ammonia. The reaction is carried out through an ion-radical mechanism that does not require passing through any barriers, therefore it might occur at low temperatures, such as on the icy mantles of meteorites. The most likely mono-substituted products are adenine and hypoxanthine, while the most likely bi-substituted products are isoguanine and guanine. In purine, we show that functional group replacement occurs in a sequential manner. Of course, nothing in our calculations suggests that other processes resulting in less energetically advantageous products are prohibitive. Other alternatives abound, but they are beyond the scope of this investigation.
Hypoxanthine (also called d-Inosine) has been shown to preferentially base pair with cytosine, indicating that it can act as a guanine surrogate. In addition, a kind of RNA editing in cells transforms adenosine to inosine (hypoxanthine) in order to take use of cytosine's base pairing abilities. These findings suggest that, despite not being one of the biological bases found in DNA and RNA, hypoxanthine may have played a crucial role in prebiotic chemistry. Our findings demonstrate how hypoxanthine, as well as the nucleobases adenine and guanine, may be easily produced in irradiation ices if purine is present.
Conclusions
Purine was used as a dopant in mixed 10:1 water-ammonia molecular ices, and we investigated the variety of products that could emerge from hydroxyl and amino group substitutions in purine. We discovered that both amino and hydroxyl group replacements are energetically beneficial processes, with the 2 and 6 locations in the six-membered cyclic ring being the most favoured places for both functional group substitutions based on quantum chemical calculations. The energy of amine group substitutions were marginally lower than those of hydroxyl group substitutions, implying that amino group substituted products may have a minor advantage over hydroxyl group substituted products. Nonetheless, larger yields of 2-hydroxypurine and hypoxanthine over adenine, and xanthine over 2,6-diaminopurine, should result from the higher abundance of hydroxyl. As observed in the bottom right corner of Fig, starting with 2-hydroxypurine, an amine group substitution should lead to the preferential production of isoguanine over guanine. Following the upper-left corner of Fig, starting with adenine, should produce isoguanine. An amino group substitution from hypoxanthine to guanine would result in guanine. This was substantiated by the experimental finding that despite NH3 and H2O being present in a 1:10 ratio, the mono-amino substituted product adenine and the mono-hydroxylated product hypoxanthine were discovered to be in 1:4 ratios. In comparison to bi-substituted compounds, mono-substituted compounds were almost an order of magnitude more common in experimental samples. The quantity of the product is also reflected in the bi-amino and bi-hydroxy purine ratios. This is primarily owing to the sequential pattern of the substitutions, which reinforces our sequential reaction mechanisms.
In the photo-processing of purine in ices, H2O serves as a reactant, a catalyst, and a solvent, among other things. H2O produces hydroxyl radicals, which are involved in hydroxylation processes. H2O serves as a catalyst in the cationic mechanism's substitution reactions, abstracting a proton to produce products. Furthermore, the H2O matrix is required not just to catch protons produced as a by-product of the reaction, but also to allow the reactant cations to survive. Anions, rather than cations, are formed in the absence of H2O, i.e., in pure NH3 ices, which stymies the processes. The experimental finding of no products in pure ammonia ices demonstrates these critical roles of H2O.
Condensed phase calculations demonstrate the importance of the water matrix in amination and hydroxylation. In some situations, one H2O molecule was sufficient to initiate the process, but in most situations, more than one H2O molecule, i.e., an H2O matrix, is required. The reactants, intermediates, and products all become solvated, although to varying degrees, according to the C-PCM calculations. The products are better solvated than the reactants and intermediates, allowing the reactions to progress. This agrees with earlier and current gas phase computations, indicating that the reaction in the gas phase is incomplete.
We expected that adenine and 2-hydroxypurine are the most energetically advantageous mono-substituted products, and isoguanine is the most favoured bi-substituted product, based on our quantum chemistry simulations. The most abundant products in the lab are adenine and hypoxanthine (2-hydroxypurine standards were not available), followed by bi-substituted compounds. We demonstrated how the reaction proceeds to the products with the help of H2O in the presence of the H2O matrix. The cationic mechanism might be used to carry out the reaction in a pure H2O ice. However, no products are discovered in the pure NH3 media, where cations are not generated but anions are. This notion is supported by the fact that the products were not detected in pure NH3 ice in an experiment. All evidence suggests that the cationic mechanism is active in pure H2O and mixed molecular ices, in addition to the catalytic and solvation actions of H2O the matrix.
Definition
DNA and RNA are crucial in determining who you are. But what is it that makes DNA unique? Nucleotides are chemical molecules with a specific makeup that make up DNA and RNA. DNA would be the tower, and nucleotides would be the individual Legos, if you had a Lego toy tower.
As a result, a polynucleotide is a chain of nucleotides. DNA is a pair of polynucleotides, while RNA is a polynucleotide. We'll discover how nucleotides link to form a polynucleotide strand in this session.
Structure
Consider a row of flags, such as Tibetan prayer flags or the flags displayed at a car dealership. The string acts as a backbone, connecting the flags. A polynucleotide, like any other molecule, contains a backbone. We call this polynucleotide backbone a sugar-phosphate backbone because it is made up of the sugar and phosphate components of nucleotides.
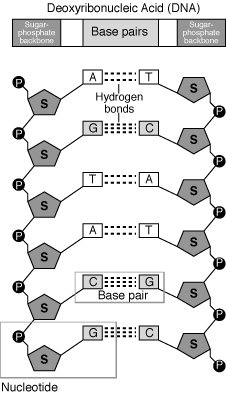
A polynucleotide strand has a sequence of bases called nitrogenous bases, just like a strand of flags can have a sequence - say, "red, red, blue, red, green..." The bases have various chemical names instead of colours. Adenine, guanine, cytosine, and thymine are the bases in DNA. This means that the sequence of a polynucleotide can look like this:
ACGTCGTATATCGTAGCTGTCAGTCGAGTAC...
RNA's polynucleotides are a little different; instead of thymine, they have uracil. They also have a different type of sugar in their backbone.
How Nucleotides Join Together
A base, a sugar, and a phosphate are all present in each nucleotide. Polymerase is an enzyme that binds the sugars to the phosphates. Each polymerase is unique to DNA and RNA.
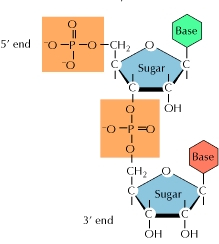
The polymerase adds additional nucleotides to the strand's bottom end, referred known as the 3' end. The phosphate group connects to the sugar's third carbon, hence the name. The phosphate group connects to the fifth carbon of the sugar at the 5' end of the strand.
One strand of a double polynucleotide, such as DNA, is upside-down with relation to the other, with its 3' end near the 5' end of the other. As a result, the two strands are known as antiparallel.
References:
1. Talwar, G.P. & Srivastava, M. Textbook of Biochemistry and Human Biology, 3rd Ed. PHI Learning.
2. Berg, J.M., Tymoczko, J.L. & Stryer, L. Biochemistry, W.H. Freeman, 2002.
3. Murray, R.K., Granner, D.K., Mayes, P.A. And Rodwell, V.W. (2009) Harper’s Illustrated Biochemistry. XXVIII edition. Lange Medical Books/ McGraw-Hill.
4. Berg, J.M., Tymoczko, J.L. And Stryer, L. (2006) Biochemistry, 6th Edition. W.H. Freeman and Co. (2002).
5. Wilson, K. & Walker, J. Practical Biochemistry. Cambridge University Press (2009).




