Unit - 2
Classical Statistics - II
I tabulate the changes in thermodynamic functions for an ideal gas as it transitions from one state to another in this section.

*It's worth noting that we can write "isothermal processes on an ideal gas" instead of "isothermal processes on a real gas.” (V2/V1) = (P1/P2).
The entries for the rise in the Helmholtz and Gibbs functions for an adiabatic process will have a difficulty in that, in order to calculate ∆A or ∆G, It appears that knowing S1 and S2, as well as their differences, is required. For the time being, this is a problem to scribble on one's shirt cuff and return to later.
An ideal gas is a useful tool for learning about the thermodynamics of a fluid with a complicated equation of state. We will summarise the traditional thermodynamics of an ideal gas with constant heat capacity in this section.
2.1.1 Internal energy
The total molecular kinetic energy contained in given amount can be calculated using the ideal gas law M = ρVof the gas becomes,
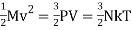 (1)
(1)
The quantity 3 comes from the fact that pointlike particles have three independent translational degrees of freedom. As a result of the preceding equations, there is an internal kinetic energy.1/2 kT Each translational degree of freedom has a number linked with it. Monatomic gases, such as Argon, have spherical molecules and thus only three translational degrees of freedom, whereas diatomic gases, such as nitrogen and oxygen, have stick-like molecules with two extra rotational degrees of freedom orthogonal to the bridge connecting the atoms, and multi-atomic gases, such as carbon dioxide and methane, have three extra rotational degrees of freedom. These degrees of freedom will also have kinetic energy, according to statistical mechanics' equipartition theorem. ½ kT as a particle Molecules also have vibrational degrees of freedom, which can be stimulated, but we'll ignore these for now. In an ideal gas, the internal energy of N particles is defined as,β
U =NkT,(2) 2
Where βis the number of degrees of freedom. Physically a gas may dissociate or even ionize when heated, and thereby change its value of β, but we shall for simplicity assume that β is in fact constant with β= 3 for monatomic, β= 5 for diatomic, and β= 6 for multiatomic gases. For mixtures of gases the number of degrees of freedom is the molar average of the degrees of freedom of the pure components.
2.1.2 Heat Capacity
Let's say we want to boost the temperature of the gas by a certain amount.δTwithout altering the volume Because no labour is done and energy is preserved, the required amount of heat is generated.δQ= δU= CVδTwhere the constant,
CV = 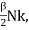 (3)
(3)
The heat capacity at constant volume is what it's termed.
If, on the other hand, the gas pressure is maintained while the temperature is increased,δT, We must also consider that the volume extends by a specific amount V, causing work to be done on the surroundings. This effort has increased the quantity of heat required.δQ= δU+ PδV.For constant pressure, we can use the ideal gas law.PδV= δ(PV ) = NkδT. As a result, the quantity of heat required per unit of temperature rise at constant pressure is
CP = CV + Nk,(4)
At constant pressure, this is referred to as the heat capacity. It is consistently larger thanCV because it entails expansionary work
2.1.3 The adiabatic index
The heat capacities' dimensionless ratio,
γ = 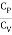 = 1+
= 1+  (5)
(5)
Is known as the adiabatic index for reasons that will be explained later. Heat capacity are usually expressed in terms of rather than.β,
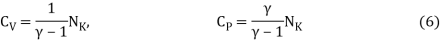
2.1.4 Entropy
The heat that must be introduced to the system in an infinitesimal operation is, when neither the volume nor the pressure are held constant.
δQ = ∂U + PδV = CVδT + NkT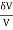 (7)
(7)
It is a mathematical fact that no function with this equation as differential exists, Q(T,V). On the other hand, it may be immediately proved (via insertion) that
δS = 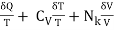 (8)
(8)
May be combined to produce a function
S = CV logT+ NklogV+ const,(9)
The entropy of the amount of ideal gas is known as the entropy of the amount of ideal gas. Entropy is only defined up to an arbitrary constant because it is an integral. The entropy of a gas, like its energy, is an abstract quantity that can't be tested physically. We can compute the value of energy and entropy in any condition since both numbers are dependent on the quantifiable thermodynamic variables that characterise the state of the gas.
2.1.5 The two fundamental laws of thermodynamics
The reason for this is that the two basic rules of thermodynamics are expressed in terms of energy and entropy. Both laws apply to processes that take place in an isolated system that is not permitted to exchange heat with the environment or conduct work on it. In an isolated system, the First Law asserts that the energy remains unchanged regardless of the process. This means that an open system's energy can only be changed by exchanging heat or work with the environment. We really used this law to derive the heat capacity and entropy indirectly. According to the Second Law, entropy cannot decrease. In the real world, an isolated system's entropy must increase. The entropy will remain constant only if all of the processes in the system are entirely reversible at all times. Reversibility is an ideal that can only be attained by extremely slow quasistatic processes made up of an endless number of microscopic reversible steps. Almost every real-world process is irreversible to some extent.
2.1.6 Isentropic processes
The term adiabatic refers to any process in an open system that does not exchange heat with the surroundings. Furthermore, since the procedure is reversible, then follows that∂Q = 0 each tiny step, to ensure that the∂S = ∂Q/T = 0. In every reversible, adiabatic process, the entropy must remain constant. For this reason, such a process is referred to as isentropic. We can write the entropy as, using the adiabatic index.S = CV log(TV γ−1) + const, (10)
From this it follows that
TV γ−1 = const,(11)
In an ideal gas for any isentropic process Using the ideal gas law to solve the problemV ∼T/P, This can be written as,
TγP1−γ= const.(12)
Rather than removing,T ∼PV ,The most common form of the isentropic state isPV γ= const. (13)
It's worth noting that the constants in these three equations differ.
2.1.7 Isothermal versus isentropic bulk modulus
The bulk modulus of a strictly isothermal ideal gas with P = nkTis equal to the pressure,

The indicator T indicates that the temperature must be kept constant while we differentiate (in the typical thermodynamic style of writing derivatives). The isentropic condition can be represented in one of three ways in terms of mass density (with three different constants),
Pρ−γ= const,Tρ1−γ= const,TγP1−γ= const.(15)
Using the first we find the isentropic bulk modulus of an ideal gas,

The index S now indicates that the entropy must be maintained constant. In all materials, the distinction between isothermal and isentropic bulk modulus is necessary, though there is little difference between the two for almost incompressible liquids.KS and KT.
The initial computation of the speed of sound in air, using essentially the ideal gas law with constant temperature, was one of Isaac Newton's greatest achievements. Because normal sound waves fluctuate so quickly, compression and expansion are fundamentally isentropic processes, his result did not match the experiment.
Sound travels at a constant pace.

√ such that the ratio between the isentropic and isothermal sound velocities is cS/cT= γ. For air with γ= 1.4 this amounts to an 18% error in the sound velocity. Much later in 1799, Laplace derived the correct value for the speed of sound.
2.1.8 Specific quantities
We always think of a macroscopic volume of matter with the same thermodynamic properties throughout the volume in classical thermodynamics. Volume, mass, energy, entropy, and specific heats are all extensive quantities, which means that the amount of any of them in a composite system is equal to the total of the parts' amounts. When a system is constructed together from its pieces, pressure, temperature, and density are contrast intensive quantities that cannot be added. An intensive amount becomes a field that varies from place to place in continuum physics, whereas an extensive quantity becomes an integral over the density of the quantity. The natural field to introduce for an extensive quantity like energy is the specific internal energy u, because a material particle with a set number of molecules has a fixed mass. u = dU/dM, which is the amount of energy per unit of mass in the neighborhood of a given point. The actual energy density becomes dU/dV= ρu, and the total energy in a volume

Temperature, pressure, and density are all examples of intense quantities. Similarly, the specific heat is defined as the local heat capacity per unit of mass, which may be written as
u = cVT = 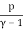 (19)
(19)
The specific heat multiplied by the absolute temperature equals the specific energy of an ideal gas.
Let's start with entropy in its most basic form, as it appears to macroscopic observers.
Entropy's behaviour can be described by comparing it to that of energy. Energy can be transmitted between systems, but it is always conserved on a global scale. Entropy, like energy, may be transported between systems but is retained in some processes. These processes are referred to as reversible because they can occur in both directions, symmetrically in time. Entropy is produced via a variety of methods (its total amount increases). Entropy, on the other hand, can never be eliminated, which is why the actions that generate it are irreversible.
This macroscopic physics approach (= human scale, or typically non-microscopic sizes; regrettably the only one offered in many thermodynamics classes) is unsatisfying because it makes the nature of entropy, its formation process, and its irreversibility look like mysteries.
Entropy has a specific definition in the fundamental laws (quantum physics without measurement) that explain elementary (microscopic) processes, but all processes are reversible, therefore this determined entropy is retained. Thus, entropy production is viewed as an emergent process that "occurs" solely in the sense that it approximates how things can be summed up in practise when vast groupings of disordered particles are involved. This approximation technique has an impact on how the system's effective states are conceived at different periods, and hence on the entropy values that are computed from these effective states. Quantum decoherence, which is normally required to qualify a process as a measurement in quantum physics, is another example of this emergent process of entropy formation. In the next pages, we'll go into the microscopic definition and genesis of entropy in greater detail.
2.2.1 Heat and temperature
Let us begin by defining temperature as a physical quantity that is positive in most cases in the correct physical sense, that is, with respect to the true physical zero value, from which the usual conventions of naming temperatures differ by an additive constant: the true physical zero value of temperature, also known as "absolute zero," corresponds to the conventional figure of -459.67 °F or -273.1 °C. Temperature in Kelvins (K) is defined as the difference between the temperatures of fusion and boiling of water at atmospheric pressure and is measured in the same unit as Celsius degrees.
Any material system growing within defined volume and energy restrictions has a maximum amount of entropy. Thermal equilibrium is a state often determined by these parameters in which a body has reached its maximum level of entropy within these boundaries (material content, volume and energy). Another significant physical property given to each system in thermal equilibrium is its temperature, which is defined as follows.
Entropy is frequently transported in conjunction with a quantity of energy. Amount of heat is a mixture of energy and entropy that can transfer from one system to another. (Energy and entropy are not separate objects that move, but rather two-substance fluids that "mix" during contact by diffusion, with only the differences in amounts on each side mattering.) Heat can come in a variety of forms, including direct touch and radiation.
Temperature is defined as the energy/entropy ratio of a given amount of heat.
The temperature of a system in thermal equilibrium is the temperature of the little amounts of heat it is ready to send to or from the outside, as it evolves between close stages of thermal equilibrium. Temperature is defined as the difference between two points (during heat exchanges as the system follows a smooth, reversible change between thermal equilibrium states),
T = δQ/dS
Where
- S = entropy (contained in the system)
- δQ = energy of received heat, = dE + PdV where E = energy, P = pressure, V = volume.
In this way, the variations dE of energy, dS of entropy and dV of volume, of a system with temperature T during (reversible) transfer of a small amount of heat at the same temperature, are related by
DE = TdS − PdV
Where TdS is the energy received from heat and − PdV is the energy received from the work of pressure.
Heat and temperature attributes can be metaphorically likened to the concept of a rubbish market with negative prices: the dictionary is
Thermodynamics | Garbage market |
Entropy | Mass of garbage |
Energy | Money |
−temperature | Negative price of garbage |
Because heat must preserve energy but can also increase entropy, it can only flow from "warm" to "cold" objects (greater temperature reduces entropy by a smaller amount for the transferred energy) (with lower temperature, getting more entropy for this energy). When this amount of heat reaches a lower-temperature object, it increases its entropy.
Any transmission of entropy between systems usually comes with a cost: it's an irreversible process that generates additional entropy. Heat flows, for example, are approximately proportional to the temperature differential between bodies near a given temperature. To make them faster, the temperature differential must increase, causing the transfer to generate more entropy. Alternatively, releasing heat causes the environment to become temporarily warmer, making the release more expensive. By slowing down the transfer, this cost can be minimised (approaching reversibility).
The fact that warm bodies convey their heat faster than cold bodies causes heat to move from the warm to the cold. So, at greater temperatures, heat transfer is faster, not just in terms of energy, but also in terms of entropy (a possibility of speed that can be traded with the fact of producing less entropy). Warmer things' radiation, in particular, has both more energy and entropy, as we'll discover below. Pure energy (which can be thought of as heat with an infinite temperature) can often be transmitted reversibly at the limit.
Entropy-creating processes in life (and machines) must transport their entropy away in order to continue working. Because this is usually only possible with energy in the form of heat, these systems must be given pure energy (or warmer energy with less entropy) in exchange. In contrast to the abundant heat energy in the environment, the purity of the received energy is what makes it useful. Still, releasing heat in a sufficient flow can be a problem, which is why power stations, for example, must be located near rivers in order to discharge their heat into the water.
2.2.2 The entropy in the Universe
For example, life on Earth is characterised by numerous irreversible processes that constantly generate entropy. The stability of this quantity around average values far below this maximum (to allow life to continue) is made possible by the continuous transfer of created entropy from Earth to outer space, in the form of infrared radiation, because there is a limit to the amount of entropy that can be contained in given limits of volume and energy (which carries quite more entropy than sunlight in proportion to its amount of energy because it is colder).
This radiation then travels over interstellar space, eventually arriving in intergalactic space. As a result, the evolution of life is fueled not only by sunlight energy (high-temperature heat), but also by the ever-larger and colder intergalactic vacuum, which the cosmic expansion supplies as a massive entropy bin. Both are complementary, in the sense that trading between two marketplaces with different pricing might result in a profit.
Even so, the entropy of visible and infrared light emitted by stars and planets is a small fraction of the total entropy in the universe. The cosmic microwave background, among electromagnetic radiations, already possesses comparable energy to visible and infrared light (1), and hence far more entropy (ignoring the entropy of practically undetectable particles: dark matter, neutrinos, gravitons...).
The enormous black holes at galaxy centres, however, account for the majority of the universe's entropy. Indeed, one of the most profoundly irreversible processes in the Universe is the fall of matter into black holes, which contributes to the growth of their size and therefore of their entropy (proportional to the area of their horizon) (that will only be "reversed" after very unreasonable times by "evaporation" in a much, much colder universe...)
2.2.3 Amounts of substance
We must relate the behaviours of temperature and entropy to other physical quantities in order to express them quantitatively. In other words, entropy levels are essentially convertible into substance levels. Let's start with an explanation of what this is.
The number of atoms or molecules included in macroscopic things is measured by the amount of substance. As a result, its deep meaning is that of natural numbers, but the unit number (one solitary atom or molecule) is too large to be meaningful.
This idea derives from science, where chemical processes require precise amounts of materials to produce molecules with the correct amount of atoms (this was first an observed fact at the beginning of the 19th century, until its explanation in terms of atoms was clearly established later that century).
The conventional unit for amounts of substance is the mol: 1 mol means NA molecules, where the number NA≈ 6.022×1023 is the Avogadro constant. Thus, n mol of some pure substance contains n×N A molecules of this substance.
This number comes from the choice that 1 mol of Carbon-12 weights 12 grams (thus roughly, 1 mol of hydrogen atoms weights 1 gram = 0.001 kg, with a slight difference due to the nuclear binding energy, converted into mass by E=mc2).
It can be seen as quantity NA ≈ 6.022×1023 mol-1.
2.2.4 Physical units of temperature and entropy
In the ideal gas law, the gas constant R=8.314 J mol−1K−1, is the natural conversion constant by which the temperature T (represented in Kelvin) is physically involved as a composite of other physical quantities in the form of the product RT. This gas constant is always present in any temperature-related phenomena, even when solids are used instead of gases, therefore temperature (and consequently entropy) has just a traditional unit, although its underlying physical nature is that of a composite of other physical qualities.
The ideal gas law, in particular, gives the physiologically valid expression of temperature, RT, as an energy per unit of substance (which explains the units involved in the value of R). It also reduces entropy (originally expressed in J/K) to a level comparable to a substance amount.
In microscopy, moles are substituted by numbers of molecules, resulting in the Boltzmann constant as the conversion factor.
k= R/NA: a temperature T microscopically appears as a typical amount of energy E=kT, while an amount of entropy S becomes a real number S/k.
This implies that changes in the temperature of a given object are proportional to the amount of heat energy exchanged.
2.2.5 Helmholtz Free energy
There are two issues with common choices for defining the Helmoltz Free energy that I could find in the literature. The first is whether it should be written with an A or a T. The question is whether the temperature T should be defined as that of the considered object or that of the environment in its defining phrase E TS. However, both functions are intriguing. So here's how we'll present them both: T stands for the object's (varying) temperature.
And T0 the (constant) temperature of the environment, let
F = E − T0 S
A = E − TS
Their variations are given by
DF = (T−T0)dS − PdV
dA = −SdT − PdV
Another remarkable function is the function A/T = E/T − S because its variations are
d(A/T) = d(E/T − S) = E d(1/T) + (dE − TdS)/T = E d(1/T) − PdV/T
The special interest of these quantities will appear when explaining the nature of entropy.
2.3.1 Entropy of Mixing, and Gibbs' Paradox
We defined the increase of entropy of a system by supposing that an infinitesimal quantity dQ of heat is added to it at temperature T, and that no irreversible work is done on the system. We then asserted that the increase of entropy of the system is dS = dQ/T. If some irreversible work is done, this has to be added to the dQ.
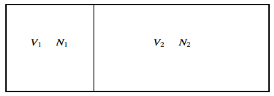
We also pointed out that any spontaneous heat transfer from one part to another part in an isolated system was likely (very likely!) to be from a hot region to a cooler region, and that this was likely (very likely!) to result in an increase in the closed system's entropy, if not the entire Universe. We analysed a box separated into two parts, one with a hot gas and the other with a cooler gas, and we explored what would happen if the wall between the portions were removed. We also examined a scenario in which the wall was used to divide two gases made up of red and blue molecules. Both scenarios appear to be pretty similar. A flow of heat is not the same as a flow of the "imponderable fluid" referred to as "caloric." Rather, it is the interaction of two groups of molecules with originally opposing qualities ("quick" and "slow," or "hot" and "cool," respectively). In either instance, spontaneous mixing, rising randomness, rising disorder, or growing entropy are likely (extremely likely!) to occur. As a result, entropy is seen as a measure of disorder. We will compute the increase in entropy when two distinct types of molecules are mixed in this section, without taking into account the passage of heat. If you study statistical mechanics, you'll notice that the concept of entropy as a measure of disorder becomes more evident. Consider a box containing two gases, separated by a partition. The pressure and temperature are the same in both compartments. The left-hand compartment contains N1 moles of gas 1, and the right-hand compartment contains N2 moles of gas 2. The Gibbs function for the system is
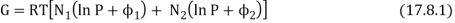
Remove the divider and wait until the gases are thoroughly mixed with no pressure or temperature change. Gas 1's partial molar Gibb’s function is

And the partial molar Gibbs function of gas 2 is

Here the pi are the partial pressures of the two gases, given by and p1 = n1P, p2 = n2P where the ni are the mole fractions.
The total Gibbs function is now N1µ1 + N2µ2, or
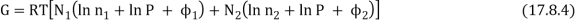
As a result, the new Gibbs function is equal to the old Gibbs function.
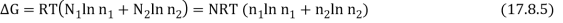
Because the mole fractions are fewer than 1, this results in a drop in the Gibbs function.
The new entropy minus the original entropy is ΔS=−[∂(ΔG)∂T]P, which is

Because the mole fractions are smaller than one, this is a good thing.
When we combine different gases, we get similar formulations for the rise in entropy.
Here's a different perspective on the same issue. (Keep in mind that the mixing is assumed to be perfect and that the temperature and pressure remain constant throughout.)
Here's the box, which is divided by a partition:
Concentrate your attention entirely upon the left-hand gas. Remove the partition. In the first nanosecond, the left-hand gas expands to increase its volume by dV, its internal energy remaining unchanged (dU = 0). The entropy of the left-hand gas therefore increases according to 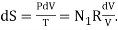 By the time it has expanded to fill the whole box, its entropy has increased. RN1 ln(V/V1). Likewise, the entropy of the right-hand gas, in expanding from volume V2 to V, has increased by RN2 ln(V/V2). Thus the entropy of the system has increased by R[ N1 ln(V/V1) ln(V/V2)], and this is equal to RN[ n1 ln(1/n1) ln(1/n2)] = − NR[n1 ln n1 + n2 ln n2].
By the time it has expanded to fill the whole box, its entropy has increased. RN1 ln(V/V1). Likewise, the entropy of the right-hand gas, in expanding from volume V2 to V, has increased by RN2 ln(V/V2). Thus the entropy of the system has increased by R[ N1 ln(V/V1) ln(V/V2)], and this is equal to RN[ n1 ln(1/n1) ln(1/n2)] = − NR[n1 ln n1 + n2 ln n2].
Where there are just two gases, n2 = 1 − n1, so we can conveniently plot a graph of the increase in the entropy versus mole fraction of gas 1, and we see, unsurprisingly, that the entropy of mixing is greatest when n1 = n2 = ½, when ∆S = NR ln 2 = 0.6931NR.
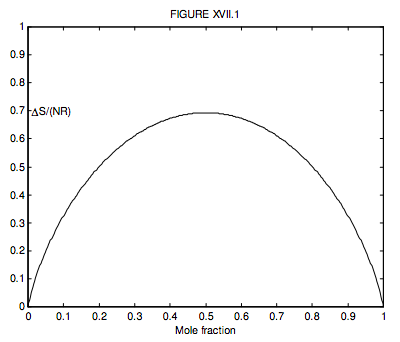
What is n1 if ΔS=12NR? (I make it n1 = 0.199 710 or, of course, 0.800 290.)
In Chapter 7, we first introduced the concept of entropy by stating that if a quantity of something changes, it becomes entropy. Heat dQ is added to a system at temperature T, the entropy increases by dS = dQ/T. We then adjusted this by pointing out that if we did some irreversible work on the system in addition to adding heat, the irreversible work was degraded to heat in any event, thus the increase in entropy was reduced dS = (dQ + dWirr)/T. We can now see that simply combining two or more gases at the same temperature causes an increase in entropy. The same holds true for combining any substance, not just gases, despite the fact that the formula is the same −NR[n1 ln n1 + n2 ln n2] Of course, this only applies to perfect gases. This was mentioned in passing in Chapter 7, but it has now been quantified. Two different gases placed together will spontaneously and possibly (very certainly!) irreversibly combine over time, increasing the entropy. It is extremely improbable that a combination of two gases will spontaneously split, lowering entropy.
When the two gases are identical, Gibbs' paradox occurs. The preceding analysis makes no distinction between two different gases being mixed together and two similar gases being mixed together. When you have two similar gases in two compartments at the same temperature and pressure, nothing changes when the partition is removed, hence the entropy should not change. This remains a conundrum within the boundaries of classical thermodynamics, which is resolved in the study of statistical mechanics.
Consider a reversible chemical reaction of the type Reactants Products, where we don't care which are the "reactants" and which are the "products." Assume that the Gibbs function of a mixture composed solely of "reactants" and no "products" is less than that of a mixture composed entirely of "products." A mixture of reactants and products will have a lower Gibbs function than either reactants or products alone. Indeed, as we progress from reactants to products, the Gibbs function will resemble the following:
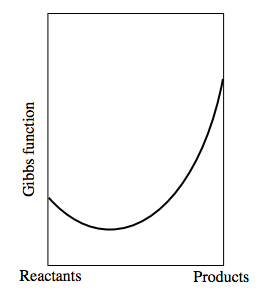
The Gibbs function of the reactants alone is shown on the left side. The Gibbs function for the products is shown on the right-hand side. When the Gibbs function is at its minimum, we have reached equilibrium.
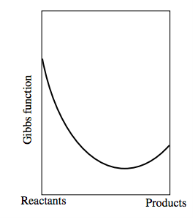
The graph would look like this if the Gibbs function of the reactants was bigger than that of the products:
Key takeaway:
- This is known as the Gibbs paradox. The dilemma can be explained by assuming that the gas particles are indistinguishable. This indicates that all states that differ only in particle permutation should be treated as the same.
- When you have two similar gases in two compartments at the same temperature and pressure, nothing changes when the partition is removed, hence the entropy should not change.
The Sackur-Tetrode equation is historically significant since it predicts quantization (and a value for Planck's constant h) based only on thermodynamic experiments. The conclusion establishes a foundation for quantum theory that is independent of Planck's original conception, which he established in the theory of blackbody radiation, and Einstein's photoelectric effect-based quantization conception.
The Sackur–Tetrode equation is a formula for the entropy of a monatomic classical ideal gas that adds quantum concepts to provide a more complete description of its validity regime. Hugo Martin Tetrode (1895–1931) and Otto Sackur (1880–1914) independently devised the Sackur–Tetrode equation around the same time in 1912 as a solution to Boltzmann's gas statistics and entropy equations.
2.4.1 Calculation of 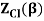
We now use the density of state for the one particle system to derive the partition function (canonical ensemble).
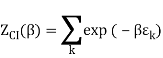
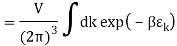
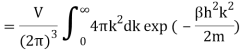
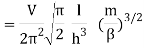
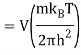
2.5.1 Law Of Equi-partition
Law of equipartition of energy: Statement
The entire energy of every dynamic system in thermal equilibrium is shared evenly among the degrees of freedom, according to the law of energy equipartition.
The kinetic energy of a single molecule is expressed as along the x-axis, y-axis, and z-axis.
1/2m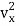 along the x-axis
along the x-axis
1/2m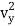 , along the y-axis
, along the y-axis
1/2m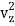 , along the z-axis
, along the z-axis
When the gas is at thermal equilibrium, the average kinetic energy is denoted as
(1/2m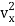 ),along the x-axis
),along the x-axis
(1/2m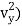 , along the y-axis
, along the y-axis
(1/2m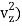 , along the z-axis
, along the z-axis
The average kinetic energy of a molecule is determined by, according to the kinetic theory of gases.
½ m vrms2 = 3/2 Kb T
Where Vrms is the root-mean-square velocity of the molecules, Kb is the Boltzmann constant and T is the temperature of the gas.
Because there are three degrees of freedom in a monoatomic gas, the average kinetic energy per degree of freedom is given by KEx = ½ KbT
If a molecule is free to move in space, it requires three coordinates to indicate its location, implying that it has three degrees of freedom in translation. It has two translational degrees of freedom if it is confined to travel in a plane, and one translational degree of freedom if it is bound to travel in a straight line. The degree of freedom in the instance of a triatomic molecule is 6. And the per-molecule kinetic energy of the gas is given as,
6 × N × ½ Kb T = 3 × R/N N Kb T = 3RT
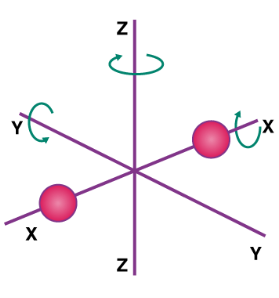
A monoatomic gas molecule, such as argon or helium, has only one translational degree of freedom. The gas's kinetic energy per molecule is provided by
3 × N × ½ Kb T = 3 × R/N N Kb T = 3/2 RT
Diatomic gases with three translational degrees of freedom, such as O2 and N2, can only spin around their centre of mass. Due to its 2-D shape, they only have two independent axes of rotation, and the third rotation is unimportant. As a result, only two rotational degrees of freedom are taken into account. As a result, the molecule has two degrees of rotation, each of which contributes a term to the total energy, which is made up of transnational and rotational energy.
2.5.2 Application of law of equi-partition energy in specific heat of a gas
Meyer’s relation CP − CV = R connects the two specific heats for one mole of an ideal gas.
Equipartition law of energy is used to calculate the value of CP − CV and the ratio between them γ = CP / CV.
Here γ is called adiabatic exponent.
i) Monatomic molecule
Average kinetic energy of a molecule
=[ 3/2 kT]
Total energy of a mole of gas
= 3/2 kT × NA = 3/2 RT
For one mole, the molar specific heat at constant volume
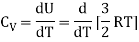
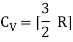
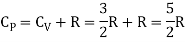
The ratio of specific heats,
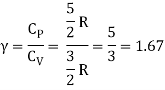
Ii) Diatomic molecule
Average kinetic energy of a diatomic molecule at low temperature = 5/2kT
Total energy of one mole of gas
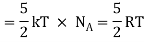
(Here, the total energy is purely kinetic)
For one mole Specific heat at constant volume
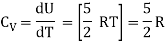
But 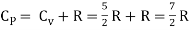
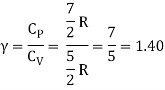
Energy of a diatomic molecule at high temperature is equal to 7/2RT
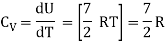
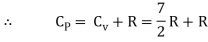
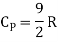
Note that the CV and CP are higher for diatomic molecules than the mono atomic molecules. It implies that to increase the temperature of diatomic gas molecules by 1°C it require more heat energy than monoatomic molecules.
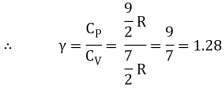
Iii) Triatomic molecule
a) Linear molecule
Energy of one mole = 7/2 kT × NA = 7/2 RT
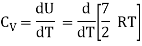
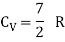
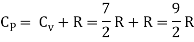
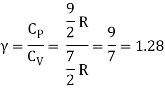
b) Non-linear molecule
Energy of a mole = 6/2 kT × NA = 6/2 RT = 3 RT
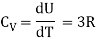
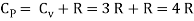
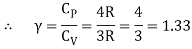
It's worth noting that the specific heat capacity of gases is temperature independent under constant volume and constant pressure, according to the kinetic theory model. However, this is not the case in reality. Temperature affects the specific heat capacity.
EXAMPLE 9.5
Find the adiabatic exponent γ for mixture of μ 1 moles of monoatomic gas and μ2 moles of a diatomic gas at normal temperature.
Solution
The specific heat of one mole of a monoatomic gas CV = 3/2 R
For μ1 mole, CV = 3/2 μ1 R CP = 5/2 μ1 R
The specific heat of one mole of a diatomic gas
CV = 5/2 R
For μ2 mole, CV = 5/2 μ2 R CP = 7/2 μ2 R
The specific heat of the mixture at constant volume CV = 3/2 μ1 R + 5/2 μ2 R
The specific heat of the mixture at constant pressure CP = 5/2 μ1 R +7/2 μ2 R
The adiabatic exponent γ = 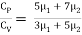
2.6.1 Partition Function for Two Level System
I have a system with Ns sites and N particles, such that Ns>>N>>1. If a site has no particle, then there is zero energy associated with that site. The N particles occupy the Ns sites and can be in energies E1 or E0, where E1>E0. We also know mm particles have energy E1 and N−mN−m have particles E0.
I'm trying to come up with the partition function for this system.
I found the total energy so far to be E = mE1 + (N – m) E0. I know the partition function is 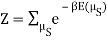 , where μs sums over all the possible microstates.
, where μs sums over all the possible microstates.
I'm trying to use the formula:
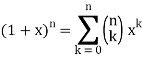
Can someone show me how this is done?
I've also figured out there are 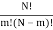 ways of getting m, E1 states and N – m, E0, states and
ways of getting m, E1 states and N – m, E0, states and 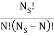 ways of arranging the N particles in the Ns sites.
ways of arranging the N particles in the Ns sites.
Following the advice of the nephente, this is what I get so far:
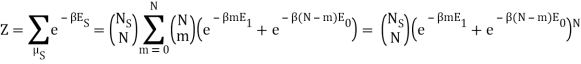
Because the particle may have energy E1 or E0, I assumed there would be a total of two exponentials. Are you suggesting that we just have:
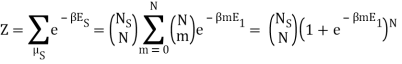
What about the zero-point energy E0? I'm having trouble figuring out which one is correct, and I'm not sure why the sum over millimetres is required. Mm was supposed to be fixed, right?
Here, I am thinking of 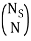 as the degeneracy for each microstate due to arranging the N particles in Ns sites.
as the degeneracy for each microstate due to arranging the N particles in Ns sites.
Edit 2:
Nephente, are you saying then that this is the correct partition function?
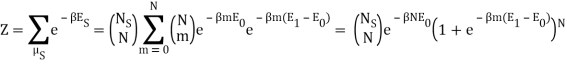
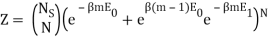
Below Absolute Zero—What Does Negative Temperature Mean?
What is the definition of negative temperature? Is it possible to create a system with a temperature below absolute zero? Can you even explain what the phrase "negative absolute temperature" means?
Yes, without a doubt. :-)
A closed system can be represented by a negative temperature under specific conditions yet, surprisingly, be hotter than the same system at any positive temperature. This article explains how everything works.
Step I: What is "Temperature"?
We need a clear definition of "temperature" to get things started. In fact, several types of "temperature" emerge in physics literature (e.g., kinetic temperature, colour temperature). The one from thermodynamics, which is in some ways the most fundamental, is pertinent here.
Our intuition tells us that if two systems in thermal contact are at the same temperature, they should exchange no heat on average. The two systems will be referred to as S1 and S2. S3 is a hybrid system that treats S1 and S2 at the same time. "How will the energy of S3 be distributed between S1 and S2?" is a crucial topic to answer in order to arrive at an usable quantitative definition of temperature. I'll discuss this quickly below, but I recommend reading K&K (linked below) for a complete, clear, and full discussion of this crucial result.
With a total energy E, S can exist in a variety of internal states (microstates). S3 atoms can share total energy in a variety of ways. Assume there are N distinct states. In the two subsystems S1 and S2, each state corresponds to a specific division of the total energy. E1 in S1 and E2 in S2 are two microstates that correspond to the same division. Only one division of the energy will occur with any substantial probability, according to a simple counting argument. It's the one containing the overwhelming majority of microstates for the entire S3 system. That figure, N(E1,E2) is just the product of the number of states allowed in each subsystem, N(E1,E2) = N1(E1)*N2(E2), and, since E1 + E2 = E, N(E1,E2) reaches a maximum when N1*N2 is stationary with respect to variations of E1 and E2 subject to the total energy constraint.
For convenience, physicists prefer to frame the question in terms of the logarithm of the number of microstates N, and call this the entropy, S. You can easily see from the above analysis that two systems are in equilibrium with one another when (dS/dE)1 = (dS/dE)2, i.e., the rate of change of entropy, S, per unit change in energy, E, must be the same for both systems. Otherwise, energy will tend to flow from one subsystem to another as S3 bounces randomly from one microstate to another, the total energy E3 being constant, as the combined system moves towards a state of maximal total entropy. We define the temperature, T, by 1/T = dS/dE, so that the equilibrium condition becomes the very simple T1 = T2.For most systems, this statistical mechanical definition of temperature corresponds to your intuitive sense of temperature. T is always positive as long as dS/dE is always positive. Adding energy to a common scenario, such as a group of free particles or particles in a harmonic oscillator potential, always increases the number of accessible microstates, and this increase accelerates as total energy grows. So, starting at 0, temperature rises with increasing energy, asymptotically approaches positive infinity as energy rises.
Step II: What is "Negative Temperature"?
The fact that entropy increases monotonically with energy is not shared by all systems. For particular ranges of energies, the number of viable microstates, or configurations, actually decreases as energy is added to the system. Consider an ideal "spin-system," which consists of N atoms with spin 1/2 on a one-dimensional wire. The atoms are unable to move freely from their wiring locations. Spin-flip is the only degree of freedom they have: a particular atom's spin can point up or down. In a strong magnetic field, the total energy of the system B, pointing down, is (N+ - N-)*uB, where u represents each atom's magnetic moment and N+ and N- represent the number of atoms with spin up and down, respectively. E is zero when half of the spins are up and half are down, according to this definition. When the majority is down, it is negative; when the majority is up, it is positive. The lowest possible energy state, all the spins pointing down, gives the system a total energy of -NuB, and temperature of absolute zero. There is only one configuration of the system at this energy, i.e., all the spins must point down. The entropy is the log of the number of microstates, so in this case is log(1) = 0. If we now add a quantum of energy, size uB, to the system, one spin is allowed to flip up. There are N possibilities, so the entropy is log(N). If we add another quantum of energy, there are a total of N(N-1)/2 allowable configurations with two spins up. The entropy is increasing quickly, and the temperature is rising as well.
However, for this system, the entropy does not go on increasing forever. There is a maximum energy, +NuB, with all spins up. There is just one microstate at this maximal energy, and the entropy is zero once more. We can enable one spin down by removing one quantum of energy from the system. There are N microstates possible at this energy. As the energy is reduced, the entropy increases. In reality, for total energy zero, i.e. half of the spins up, half down, the highest entropy occurs.
When a result, we've developed a system in which, as additional energy is added, the temperature rises to positive infinity as maximum entropy is approached, with half of all spins up. As the energy climbs toward maximum, the temperature becomes negative infinite, decreasing in magnitude until it reaches zero, but always remaining negative. It is hotter when the system has a negative temperature than when it has a positive temperature. Heat will flow from the negative-temperature system to the positive-temperature system if two copies of the system, one with positive and one with negative temperature, are placed in thermal contact.
Step III: What Does This Have to Do With the Real World?
Is this system a fascinating invention of malevolent theoretical condensed matter physicists, or can it ever be achieved in the actual world? In addition to spin, atoms always have other degrees of freedom, which makes the total energy of the system limitless upward due to the atom's translational degrees of freedom. As a result, a particle can only have negative temperature in specific degrees of freedom. It makes sense to define a collection of atoms' "spin-temperature" if one condition is met: the coupling between atomic spins and other degrees of freedom is sufficiently weak, and the coupling between atomic spins is sufficiently strong, that the timescale for energy to flow from the spins into other degrees of freedom is very large compared to the timescale for thermalization. The temperature of the spins should be discussed independently from the temperature of the atoms as a whole. Under the case of nuclear spins in a strong external magnetic field, this criterion is easily met.
Radio frequency approaches can be used to boost nuclear and electron spin systems to negative temperatures. The articles listed below, as well as the references therein, contain many experiments in negative temperature calorimetry, as well as applications of negative temperature systems as RF amplifiers, etc.
Key takeaway:
- Absolute zero, often known as zero kelvins, is a temperature of 273.15 degrees Celsius (-459.67 degrees Fahrenheit) that indicates the point on a thermometer where a system has reached its lowest potential energy, or thermal motion. However, there is a catch: absolute zero is unattainable.
- The Celsius scale, often known as centigrade, is a temperature measurement scale. The degree Celsius (°C) is the measuring unit. It is one of the world's most widely used temperature units.
- The particles stop moving and all chaos vanishes at zero kelvin (minus 273 degrees Celsius). On the Kelvin scale, nothing can be colder than absolute zero.
References:
1. Statistical Mechanics, B.K.Agarwal and Melvin Eisner (New Age Inter-national)-2013
2. Thermodynamics, Kinetic Theory and Statistical Thermodynamics: Francis W.Sears and Gerhard L. Salinger (Narosa) 1998
3. Statistical Mechanics: R.K.Pathria and Paul D. Beale (Academic Press)- 2011
Unit - 2
Classical Statistics - II
I tabulate the changes in thermodynamic functions for an ideal gas as it transitions from one state to another in this section.

*It's worth noting that we can write "isothermal processes on an ideal gas" instead of "isothermal processes on a real gas.” (V2/V1) = (P1/P2).
The entries for the rise in the Helmholtz and Gibbs functions for an adiabatic process will have a difficulty in that, in order to calculate ∆A or ∆G, It appears that knowing S1 and S2, as well as their differences, is required. For the time being, this is a problem to scribble on one's shirt cuff and return to later.
An ideal gas is a useful tool for learning about the thermodynamics of a fluid with a complicated equation of state. We will summarise the traditional thermodynamics of an ideal gas with constant heat capacity in this section.
2.1.1 Internal energy
The total molecular kinetic energy contained in given amount can be calculated using the ideal gas law M = ρVof the gas becomes,
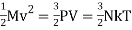 (1)
(1)
The quantity 3 comes from the fact that pointlike particles have three independent translational degrees of freedom. As a result of the preceding equations, there is an internal kinetic energy.1/2 kT Each translational degree of freedom has a number linked with it. Monatomic gases, such as Argon, have spherical molecules and thus only three translational degrees of freedom, whereas diatomic gases, such as nitrogen and oxygen, have stick-like molecules with two extra rotational degrees of freedom orthogonal to the bridge connecting the atoms, and multi-atomic gases, such as carbon dioxide and methane, have three extra rotational degrees of freedom. These degrees of freedom will also have kinetic energy, according to statistical mechanics' equipartition theorem. ½ kT as a particle Molecules also have vibrational degrees of freedom, which can be stimulated, but we'll ignore these for now. In an ideal gas, the internal energy of N particles is defined as,β
U =NkT,(2) 2
Where βis the number of degrees of freedom. Physically a gas may dissociate or even ionize when heated, and thereby change its value of β, but we shall for simplicity assume that β is in fact constant with β= 3 for monatomic, β= 5 for diatomic, and β= 6 for multiatomic gases. For mixtures of gases the number of degrees of freedom is the molar average of the degrees of freedom of the pure components.
2.1.2 Heat Capacity
Let's say we want to boost the temperature of the gas by a certain amount.δTwithout altering the volume Because no labour is done and energy is preserved, the required amount of heat is generated.δQ= δU= CVδTwhere the constant,
CV = 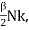 (3)
(3)
The heat capacity at constant volume is what it's termed.
If, on the other hand, the gas pressure is maintained while the temperature is increased,δT, We must also consider that the volume extends by a specific amount V, causing work to be done on the surroundings. This effort has increased the quantity of heat required.δQ= δU+ PδV.For constant pressure, we can use the ideal gas law.PδV= δ(PV ) = NkδT. As a result, the quantity of heat required per unit of temperature rise at constant pressure is
CP = CV + Nk,(4)
At constant pressure, this is referred to as the heat capacity. It is consistently larger thanCV because it entails expansionary work
2.1.3 The adiabatic index
The heat capacities' dimensionless ratio,
γ = 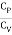 = 1+
= 1+  (5)
(5)
Is known as the adiabatic index for reasons that will be explained later. Heat capacity are usually expressed in terms of rather than.β,
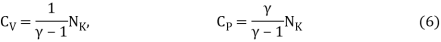
2.1.4 Entropy
The heat that must be introduced to the system in an infinitesimal operation is, when neither the volume nor the pressure are held constant.
δQ = ∂U + PδV = CVδT + NkT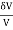 (7)
(7)
It is a mathematical fact that no function with this equation as differential exists, Q(T,V). On the other hand, it may be immediately proved (via insertion) that
δS = 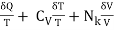 (8)
(8)
May be combined to produce a function
S = CV logT+ NklogV+ const,(9)
The entropy of the amount of ideal gas is known as the entropy of the amount of ideal gas. Entropy is only defined up to an arbitrary constant because it is an integral. The entropy of a gas, like its energy, is an abstract quantity that can't be tested physically. We can compute the value of energy and entropy in any condition since both numbers are dependent on the quantifiable thermodynamic variables that characterise the state of the gas.
2.1.5 The two fundamental laws of thermodynamics
The reason for this is that the two basic rules of thermodynamics are expressed in terms of energy and entropy. Both laws apply to processes that take place in an isolated system that is not permitted to exchange heat with the environment or conduct work on it. In an isolated system, the First Law asserts that the energy remains unchanged regardless of the process. This means that an open system's energy can only be changed by exchanging heat or work with the environment. We really used this law to derive the heat capacity and entropy indirectly. According to the Second Law, entropy cannot decrease. In the real world, an isolated system's entropy must increase. The entropy will remain constant only if all of the processes in the system are entirely reversible at all times. Reversibility is an ideal that can only be attained by extremely slow quasistatic processes made up of an endless number of microscopic reversible steps. Almost every real-world process is irreversible to some extent.
2.1.6 Isentropic processes
The term adiabatic refers to any process in an open system that does not exchange heat with the surroundings. Furthermore, since the procedure is reversible, then follows that∂Q = 0 each tiny step, to ensure that the∂S = ∂Q/T = 0. In every reversible, adiabatic process, the entropy must remain constant. For this reason, such a process is referred to as isentropic. We can write the entropy as, using the adiabatic index.S = CV log(TV γ−1) + const, (10)
From this it follows that
TV γ−1 = const,(11)
In an ideal gas for any isentropic process Using the ideal gas law to solve the problemV ∼T/P, This can be written as,
TγP1−γ= const.(12)
Rather than removing,T ∼PV ,The most common form of the isentropic state isPV γ= const. (13)
It's worth noting that the constants in these three equations differ.
2.1.7 Isothermal versus isentropic bulk modulus
The bulk modulus of a strictly isothermal ideal gas with P = nkTis equal to the pressure,

The indicator T indicates that the temperature must be kept constant while we differentiate (in the typical thermodynamic style of writing derivatives). The isentropic condition can be represented in one of three ways in terms of mass density (with three different constants),
Pρ−γ= const,Tρ1−γ= const,TγP1−γ= const.(15)
Using the first we find the isentropic bulk modulus of an ideal gas,

The index S now indicates that the entropy must be maintained constant. In all materials, the distinction between isothermal and isentropic bulk modulus is necessary, though there is little difference between the two for almost incompressible liquids.KS and KT.
The initial computation of the speed of sound in air, using essentially the ideal gas law with constant temperature, was one of Isaac Newton's greatest achievements. Because normal sound waves fluctuate so quickly, compression and expansion are fundamentally isentropic processes, his result did not match the experiment.
Sound travels at a constant pace.

√ such that the ratio between the isentropic and isothermal sound velocities is cS/cT= γ. For air with γ= 1.4 this amounts to an 18% error in the sound velocity. Much later in 1799, Laplace derived the correct value for the speed of sound.
2.1.8 Specific quantities
We always think of a macroscopic volume of matter with the same thermodynamic properties throughout the volume in classical thermodynamics. Volume, mass, energy, entropy, and specific heats are all extensive quantities, which means that the amount of any of them in a composite system is equal to the total of the parts' amounts. When a system is constructed together from its pieces, pressure, temperature, and density are contrast intensive quantities that cannot be added. An intensive amount becomes a field that varies from place to place in continuum physics, whereas an extensive quantity becomes an integral over the density of the quantity. The natural field to introduce for an extensive quantity like energy is the specific internal energy u, because a material particle with a set number of molecules has a fixed mass. u = dU/dM, which is the amount of energy per unit of mass in the neighborhood of a given point. The actual energy density becomes dU/dV= ρu, and the total energy in a volume

Temperature, pressure, and density are all examples of intense quantities. Similarly, the specific heat is defined as the local heat capacity per unit of mass, which may be written as
u = cVT = 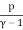 (19)
(19)
The specific heat multiplied by the absolute temperature equals the specific energy of an ideal gas.
Let's start with entropy in its most basic form, as it appears to macroscopic observers.
Entropy's behaviour can be described by comparing it to that of energy. Energy can be transmitted between systems, but it is always conserved on a global scale. Entropy, like energy, may be transported between systems but is retained in some processes. These processes are referred to as reversible because they can occur in both directions, symmetrically in time. Entropy is produced via a variety of methods (its total amount increases). Entropy, on the other hand, can never be eliminated, which is why the actions that generate it are irreversible.
This macroscopic physics approach (= human scale, or typically non-microscopic sizes; regrettably the only one offered in many thermodynamics classes) is unsatisfying because it makes the nature of entropy, its formation process, and its irreversibility look like mysteries.
Entropy has a specific definition in the fundamental laws (quantum physics without measurement) that explain elementary (microscopic) processes, but all processes are reversible, therefore this determined entropy is retained. Thus, entropy production is viewed as an emergent process that "occurs" solely in the sense that it approximates how things can be summed up in practise when vast groupings of disordered particles are involved. This approximation technique has an impact on how the system's effective states are conceived at different periods, and hence on the entropy values that are computed from these effective states. Quantum decoherence, which is normally required to qualify a process as a measurement in quantum physics, is another example of this emergent process of entropy formation. In the next pages, we'll go into the microscopic definition and genesis of entropy in greater detail.
2.2.1 Heat and temperature
Let us begin by defining temperature as a physical quantity that is positive in most cases in the correct physical sense, that is, with respect to the true physical zero value, from which the usual conventions of naming temperatures differ by an additive constant: the true physical zero value of temperature, also known as "absolute zero," corresponds to the conventional figure of -459.67 °F or -273.1 °C. Temperature in Kelvins (K) is defined as the difference between the temperatures of fusion and boiling of water at atmospheric pressure and is measured in the same unit as Celsius degrees.
Any material system growing within defined volume and energy restrictions has a maximum amount of entropy. Thermal equilibrium is a state often determined by these parameters in which a body has reached its maximum level of entropy within these boundaries (material content, volume and energy). Another significant physical property given to each system in thermal equilibrium is its temperature, which is defined as follows.
Entropy is frequently transported in conjunction with a quantity of energy. Amount of heat is a mixture of energy and entropy that can transfer from one system to another. (Energy and entropy are not separate objects that move, but rather two-substance fluids that "mix" during contact by diffusion, with only the differences in amounts on each side mattering.) Heat can come in a variety of forms, including direct touch and radiation.
Temperature is defined as the energy/entropy ratio of a given amount of heat.
The temperature of a system in thermal equilibrium is the temperature of the little amounts of heat it is ready to send to or from the outside, as it evolves between close stages of thermal equilibrium. Temperature is defined as the difference between two points (during heat exchanges as the system follows a smooth, reversible change between thermal equilibrium states),
T = δQ/dS
Where
- S = entropy (contained in the system)
- δQ = energy of received heat, = dE + PdV where E = energy, P = pressure, V = volume.
In this way, the variations dE of energy, dS of entropy and dV of volume, of a system with temperature T during (reversible) transfer of a small amount of heat at the same temperature, are related by
DE = TdS − PdV
Where TdS is the energy received from heat and − PdV is the energy received from the work of pressure.
Heat and temperature attributes can be metaphorically likened to the concept of a rubbish market with negative prices: the dictionary is
Thermodynamics | Garbage market |
Entropy | Mass of garbage |
Energy | Money |
−temperature | Negative price of garbage |
Because heat must preserve energy but can also increase entropy, it can only flow from "warm" to "cold" objects (greater temperature reduces entropy by a smaller amount for the transferred energy) (with lower temperature, getting more entropy for this energy). When this amount of heat reaches a lower-temperature object, it increases its entropy.
Any transmission of entropy between systems usually comes with a cost: it's an irreversible process that generates additional entropy. Heat flows, for example, are approximately proportional to the temperature differential between bodies near a given temperature. To make them faster, the temperature differential must increase, causing the transfer to generate more entropy. Alternatively, releasing heat causes the environment to become temporarily warmer, making the release more expensive. By slowing down the transfer, this cost can be minimised (approaching reversibility).
The fact that warm bodies convey their heat faster than cold bodies causes heat to move from the warm to the cold. So, at greater temperatures, heat transfer is faster, not just in terms of energy, but also in terms of entropy (a possibility of speed that can be traded with the fact of producing less entropy). Warmer things' radiation, in particular, has both more energy and entropy, as we'll discover below. Pure energy (which can be thought of as heat with an infinite temperature) can often be transmitted reversibly at the limit.
Entropy-creating processes in life (and machines) must transport their entropy away in order to continue working. Because this is usually only possible with energy in the form of heat, these systems must be given pure energy (or warmer energy with less entropy) in exchange. In contrast to the abundant heat energy in the environment, the purity of the received energy is what makes it useful. Still, releasing heat in a sufficient flow can be a problem, which is why power stations, for example, must be located near rivers in order to discharge their heat into the water.
2.2.2 The entropy in the Universe
For example, life on Earth is characterised by numerous irreversible processes that constantly generate entropy. The stability of this quantity around average values far below this maximum (to allow life to continue) is made possible by the continuous transfer of created entropy from Earth to outer space, in the form of infrared radiation, because there is a limit to the amount of entropy that can be contained in given limits of volume and energy (which carries quite more entropy than sunlight in proportion to its amount of energy because it is colder).
This radiation then travels over interstellar space, eventually arriving in intergalactic space. As a result, the evolution of life is fueled not only by sunlight energy (high-temperature heat), but also by the ever-larger and colder intergalactic vacuum, which the cosmic expansion supplies as a massive entropy bin. Both are complementary, in the sense that trading between two marketplaces with different pricing might result in a profit.
Even so, the entropy of visible and infrared light emitted by stars and planets is a small fraction of the total entropy in the universe. The cosmic microwave background, among electromagnetic radiations, already possesses comparable energy to visible and infrared light (1), and hence far more entropy (ignoring the entropy of practically undetectable particles: dark matter, neutrinos, gravitons...).
The enormous black holes at galaxy centres, however, account for the majority of the universe's entropy. Indeed, one of the most profoundly irreversible processes in the Universe is the fall of matter into black holes, which contributes to the growth of their size and therefore of their entropy (proportional to the area of their horizon) (that will only be "reversed" after very unreasonable times by "evaporation" in a much, much colder universe...)
2.2.3 Amounts of substance
We must relate the behaviours of temperature and entropy to other physical quantities in order to express them quantitatively. In other words, entropy levels are essentially convertible into substance levels. Let's start with an explanation of what this is.
The number of atoms or molecules included in macroscopic things is measured by the amount of substance. As a result, its deep meaning is that of natural numbers, but the unit number (one solitary atom or molecule) is too large to be meaningful.
This idea derives from science, where chemical processes require precise amounts of materials to produce molecules with the correct amount of atoms (this was first an observed fact at the beginning of the 19th century, until its explanation in terms of atoms was clearly established later that century).
The conventional unit for amounts of substance is the mol: 1 mol means NA molecules, where the number NA≈ 6.022×1023 is the Avogadro constant. Thus, n mol of some pure substance contains n×N A molecules of this substance.
This number comes from the choice that 1 mol of Carbon-12 weights 12 grams (thus roughly, 1 mol of hydrogen atoms weights 1 gram = 0.001 kg, with a slight difference due to the nuclear binding energy, converted into mass by E=mc2).
It can be seen as quantity NA ≈ 6.022×1023 mol-1.
2.2.4 Physical units of temperature and entropy
In the ideal gas law, the gas constant R=8.314 J mol−1K−1, is the natural conversion constant by which the temperature T (represented in Kelvin) is physically involved as a composite of other physical quantities in the form of the product RT. This gas constant is always present in any temperature-related phenomena, even when solids are used instead of gases, therefore temperature (and consequently entropy) has just a traditional unit, although its underlying physical nature is that of a composite of other physical qualities.
The ideal gas law, in particular, gives the physiologically valid expression of temperature, RT, as an energy per unit of substance (which explains the units involved in the value of R). It also reduces entropy (originally expressed in J/K) to a level comparable to a substance amount.
In microscopy, moles are substituted by numbers of molecules, resulting in the Boltzmann constant as the conversion factor.
k= R/NA: a temperature T microscopically appears as a typical amount of energy E=kT, while an amount of entropy S becomes a real number S/k.
This implies that changes in the temperature of a given object are proportional to the amount of heat energy exchanged.
2.2.5 Helmholtz Free energy
There are two issues with common choices for defining the Helmoltz Free energy that I could find in the literature. The first is whether it should be written with an A or a T. The question is whether the temperature T should be defined as that of the considered object or that of the environment in its defining phrase E TS. However, both functions are intriguing. So here's how we'll present them both: T stands for the object's (varying) temperature.
And T0 the (constant) temperature of the environment, let
F = E − T0 S
A = E − TS
Their variations are given by
DF = (T−T0)dS − PdV
dA = −SdT − PdV
Another remarkable function is the function A/T = E/T − S because its variations are
d(A/T) = d(E/T − S) = E d(1/T) + (dE − TdS)/T = E d(1/T) − PdV/T
The special interest of these quantities will appear when explaining the nature of entropy.
2.3.1 Entropy of Mixing, and Gibbs' Paradox
We defined the increase of entropy of a system by supposing that an infinitesimal quantity dQ of heat is added to it at temperature T, and that no irreversible work is done on the system. We then asserted that the increase of entropy of the system is dS = dQ/T. If some irreversible work is done, this has to be added to the dQ.
We also pointed out that any spontaneous heat transfer from one part to another part in an isolated system was likely (very likely!) to be from a hot region to a cooler region, and that this was likely (very likely!) to result in an increase in the closed system's entropy, if not the entire Universe. We analysed a box separated into two parts, one with a hot gas and the other with a cooler gas, and we explored what would happen if the wall between the portions were removed. We also examined a scenario in which the wall was used to divide two gases made up of red and blue molecules. Both scenarios appear to be pretty similar. A flow of heat is not the same as a flow of the "imponderable fluid" referred to as "caloric." Rather, it is the interaction of two groups of molecules with originally opposing qualities ("quick" and "slow," or "hot" and "cool," respectively). In either instance, spontaneous mixing, rising randomness, rising disorder, or growing entropy are likely (extremely likely!) to occur. As a result, entropy is seen as a measure of disorder. We will compute the increase in entropy when two distinct types of molecules are mixed in this section, without taking into account the passage of heat. If you study statistical mechanics, you'll notice that the concept of entropy as a measure of disorder becomes more evident. Consider a box containing two gases, separated by a partition. The pressure and temperature are the same in both compartments. The left-hand compartment contains N1 moles of gas 1, and the right-hand compartment contains N2 moles of gas 2. The Gibbs function for the system is
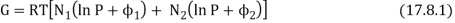
Remove the divider and wait until the gases are thoroughly mixed with no pressure or temperature change. Gas 1's partial molar Gibb’s function is

And the partial molar Gibbs function of gas 2 is

Here the pi are the partial pressures of the two gases, given by and p1 = n1P, p2 = n2P where the ni are the mole fractions.
The total Gibbs function is now N1µ1 + N2µ2, or
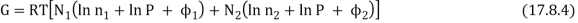
As a result, the new Gibbs function is equal to the old Gibbs function.
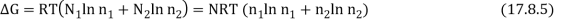
Because the mole fractions are fewer than 1, this results in a drop in the Gibbs function.
The new entropy minus the original entropy is ΔS=−[∂(ΔG)∂T]P, which is

Because the mole fractions are smaller than one, this is a good thing.
When we combine different gases, we get similar formulations for the rise in entropy.
Here's a different perspective on the same issue. (Keep in mind that the mixing is assumed to be perfect and that the temperature and pressure remain constant throughout.)
Here's the box, which is divided by a partition:
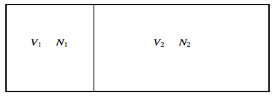
Concentrate your attention entirely upon the left-hand gas. Remove the partition. In the first nanosecond, the left-hand gas expands to increase its volume by dV, its internal energy remaining unchanged (dU = 0). The entropy of the left-hand gas therefore increases according to 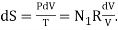 By the time it has expanded to fill the whole box, its entropy has increased. RN1 ln(V/V1). Likewise, the entropy of the right-hand gas, in expanding from volume V2 to V, has increased by RN2 ln(V/V2). Thus the entropy of the system has increased by R[ N1 ln(V/V1) ln(V/V2)], and this is equal to RN[ n1 ln(1/n1) ln(1/n2)] = − NR[n1 ln n1 + n2 ln n2].
By the time it has expanded to fill the whole box, its entropy has increased. RN1 ln(V/V1). Likewise, the entropy of the right-hand gas, in expanding from volume V2 to V, has increased by RN2 ln(V/V2). Thus the entropy of the system has increased by R[ N1 ln(V/V1) ln(V/V2)], and this is equal to RN[ n1 ln(1/n1) ln(1/n2)] = − NR[n1 ln n1 + n2 ln n2].
Where there are just two gases, n2 = 1 − n1, so we can conveniently plot a graph of the increase in the entropy versus mole fraction of gas 1, and we see, unsurprisingly, that the entropy of mixing is greatest when n1 = n2 = ½, when ∆S = NR ln 2 = 0.6931NR.
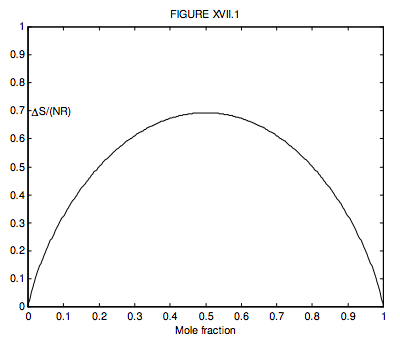
What is n1 if ΔS=12NR? (I make it n1 = 0.199 710 or, of course, 0.800 290.)
In Chapter 7, we first introduced the concept of entropy by stating that if a quantity of something changes, it becomes entropy. Heat dQ is added to a system at temperature T, the entropy increases by dS = dQ/T. We then adjusted this by pointing out that if we did some irreversible work on the system in addition to adding heat, the irreversible work was degraded to heat in any event, thus the increase in entropy was reduced dS = (dQ + dWirr)/T. We can now see that simply combining two or more gases at the same temperature causes an increase in entropy. The same holds true for combining any substance, not just gases, despite the fact that the formula is the same −NR[n1 ln n1 + n2 ln n2] Of course, this only applies to perfect gases. This was mentioned in passing in Chapter 7, but it has now been quantified. Two different gases placed together will spontaneously and possibly (very certainly!) irreversibly combine over time, increasing the entropy. It is extremely improbable that a combination of two gases will spontaneously split, lowering entropy.
When the two gases are identical, Gibbs' paradox occurs. The preceding analysis makes no distinction between two different gases being mixed together and two similar gases being mixed together. When you have two similar gases in two compartments at the same temperature and pressure, nothing changes when the partition is removed, hence the entropy should not change. This remains a conundrum within the boundaries of classical thermodynamics, which is resolved in the study of statistical mechanics.
Consider a reversible chemical reaction of the type Reactants Products, where we don't care which are the "reactants" and which are the "products." Assume that the Gibbs function of a mixture composed solely of "reactants" and no "products" is less than that of a mixture composed entirely of "products." A mixture of reactants and products will have a lower Gibbs function than either reactants or products alone. Indeed, as we progress from reactants to products, the Gibbs function will resemble the following:
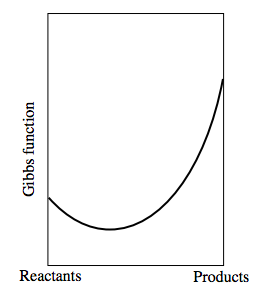
The Gibbs function of the reactants alone is shown on the left side. The Gibbs function for the products is shown on the right-hand side. When the Gibbs function is at its minimum, we have reached equilibrium.
The graph would look like this if the Gibbs function of the reactants was bigger than that of the products:
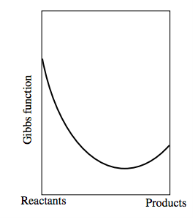
Key takeaway:
- This is known as the Gibbs paradox. The dilemma can be explained by assuming that the gas particles are indistinguishable. This indicates that all states that differ only in particle permutation should be treated as the same.
- When you have two similar gases in two compartments at the same temperature and pressure, nothing changes when the partition is removed, hence the entropy should not change.
The Sackur-Tetrode equation is historically significant since it predicts quantization (and a value for Planck's constant h) based only on thermodynamic experiments. The conclusion establishes a foundation for quantum theory that is independent of Planck's original conception, which he established in the theory of blackbody radiation, and Einstein's photoelectric effect-based quantization conception.
The Sackur–Tetrode equation is a formula for the entropy of a monatomic classical ideal gas that adds quantum concepts to provide a more complete description of its validity regime. Hugo Martin Tetrode (1895–1931) and Otto Sackur (1880–1914) independently devised the Sackur–Tetrode equation around the same time in 1912 as a solution to Boltzmann's gas statistics and entropy equations.
2.4.1 Calculation of 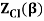
We now use the density of state for the one particle system to derive the partition function (canonical ensemble).
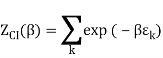
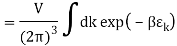
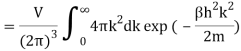
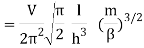
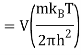
2.5.1 Law Of Equi-partition
Law of equipartition of energy: Statement
The entire energy of every dynamic system in thermal equilibrium is shared evenly among the degrees of freedom, according to the law of energy equipartition.
The kinetic energy of a single molecule is expressed as along the x-axis, y-axis, and z-axis.
1/2m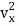 along the x-axis
along the x-axis
1/2m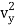 , along the y-axis
, along the y-axis
1/2m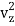 , along the z-axis
, along the z-axis
When the gas is at thermal equilibrium, the average kinetic energy is denoted as
(1/2m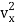 ),along the x-axis
),along the x-axis
(1/2m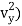 , along the y-axis
, along the y-axis
(1/2m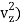 , along the z-axis
, along the z-axis
The average kinetic energy of a molecule is determined by, according to the kinetic theory of gases.
½ m vrms2 = 3/2 Kb T
Where Vrms is the root-mean-square velocity of the molecules, Kb is the Boltzmann constant and T is the temperature of the gas.
Because there are three degrees of freedom in a monoatomic gas, the average kinetic energy per degree of freedom is given by KEx = ½ KbT
If a molecule is free to move in space, it requires three coordinates to indicate its location, implying that it has three degrees of freedom in translation. It has two translational degrees of freedom if it is confined to travel in a plane, and one translational degree of freedom if it is bound to travel in a straight line. The degree of freedom in the instance of a triatomic molecule is 6. And the per-molecule kinetic energy of the gas is given as,
6 × N × ½ Kb T = 3 × R/N N Kb T = 3RT
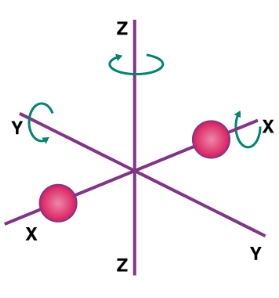
A monoatomic gas molecule, such as argon or helium, has only one translational degree of freedom. The gas's kinetic energy per molecule is provided by
3 × N × ½ Kb T = 3 × R/N N Kb T = 3/2 RT
Diatomic gases with three translational degrees of freedom, such as O2 and N2, can only spin around their centre of mass. Due to its 2-D shape, they only have two independent axes of rotation, and the third rotation is unimportant. As a result, only two rotational degrees of freedom are taken into account. As a result, the molecule has two degrees of rotation, each of which contributes a term to the total energy, which is made up of transnational and rotational energy.
2.5.2 Application of law of equi-partition energy in specific heat of a gas
Meyer’s relation CP − CV = R connects the two specific heats for one mole of an ideal gas.
Equipartition law of energy is used to calculate the value of CP − CV and the ratio between them γ = CP / CV.
Here γ is called adiabatic exponent.
i) Monatomic molecule
Average kinetic energy of a molecule
=[ 3/2 kT]
Total energy of a mole of gas
= 3/2 kT × NA = 3/2 RT
For one mole, the molar specific heat at constant volume
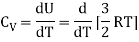
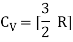
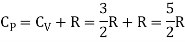
The ratio of specific heats,
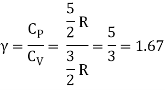
Ii) Diatomic molecule
Average kinetic energy of a diatomic molecule at low temperature = 5/2kT
Total energy of one mole of gas
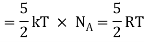
(Here, the total energy is purely kinetic)
For one mole Specific heat at constant volume
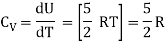
But 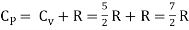
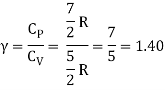
Energy of a diatomic molecule at high temperature is equal to 7/2RT
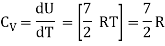
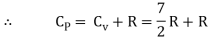
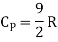
Note that the CV and CP are higher for diatomic molecules than the mono atomic molecules. It implies that to increase the temperature of diatomic gas molecules by 1°C it require more heat energy than monoatomic molecules.
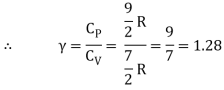
Iii) Triatomic molecule
a) Linear molecule
Energy of one mole = 7/2 kT × NA = 7/2 RT
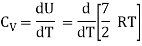
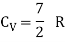
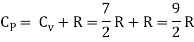
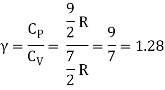
b) Non-linear molecule
Energy of a mole = 6/2 kT × NA = 6/2 RT = 3 RT
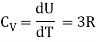
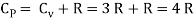
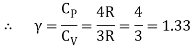
It's worth noting that the specific heat capacity of gases is temperature independent under constant volume and constant pressure, according to the kinetic theory model. However, this is not the case in reality. Temperature affects the specific heat capacity.
EXAMPLE 9.5
Find the adiabatic exponent γ for mixture of μ 1 moles of monoatomic gas and μ2 moles of a diatomic gas at normal temperature.
Solution
The specific heat of one mole of a monoatomic gas CV = 3/2 R
For μ1 mole, CV = 3/2 μ1 R CP = 5/2 μ1 R
The specific heat of one mole of a diatomic gas
CV = 5/2 R
For μ2 mole, CV = 5/2 μ2 R CP = 7/2 μ2 R
The specific heat of the mixture at constant volume CV = 3/2 μ1 R + 5/2 μ2 R
The specific heat of the mixture at constant pressure CP = 5/2 μ1 R +7/2 μ2 R
The adiabatic exponent γ = 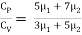
2.6.1 Partition Function for Two Level System
I have a system with Ns sites and N particles, such that Ns>>N>>1. If a site has no particle, then there is zero energy associated with that site. The N particles occupy the Ns sites and can be in energies E1 or E0, where E1>E0. We also know mm particles have energy E1 and N−mN−m have particles E0.
I'm trying to come up with the partition function for this system.
I found the total energy so far to be E = mE1 + (N – m) E0. I know the partition function is 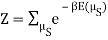 , where μs sums over all the possible microstates.
, where μs sums over all the possible microstates.
I'm trying to use the formula:
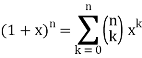
Can someone show me how this is done?
I've also figured out there are 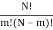 ways of getting m, E1 states and N – m, E0, states and
ways of getting m, E1 states and N – m, E0, states and 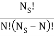 ways of arranging the N particles in the Ns sites.
ways of arranging the N particles in the Ns sites.
Following the advice of the nephente, this is what I get so far:
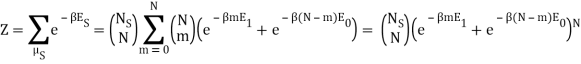
Because the particle may have energy E1 or E0, I assumed there would be a total of two exponentials. Are you suggesting that we just have:
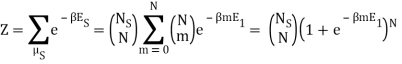
What about the zero-point energy E0? I'm having trouble figuring out which one is correct, and I'm not sure why the sum over millimetres is required. Mm was supposed to be fixed, right?
Here, I am thinking of 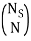 as the degeneracy for each microstate due to arranging the N particles in Ns sites.
as the degeneracy for each microstate due to arranging the N particles in Ns sites.
Edit 2:
Nephente, are you saying then that this is the correct partition function?
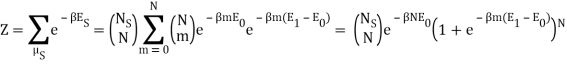
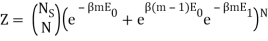
Below Absolute Zero—What Does Negative Temperature Mean?
What is the definition of negative temperature? Is it possible to create a system with a temperature below absolute zero? Can you even explain what the phrase "negative absolute temperature" means?
Yes, without a doubt. :-)
A closed system can be represented by a negative temperature under specific conditions yet, surprisingly, be hotter than the same system at any positive temperature. This article explains how everything works.
Step I: What is "Temperature"?
We need a clear definition of "temperature" to get things started. In fact, several types of "temperature" emerge in physics literature (e.g., kinetic temperature, colour temperature). The one from thermodynamics, which is in some ways the most fundamental, is pertinent here.
Our intuition tells us that if two systems in thermal contact are at the same temperature, they should exchange no heat on average. The two systems will be referred to as S1 and S2. S3 is a hybrid system that treats S1 and S2 at the same time. "How will the energy of S3 be distributed between S1 and S2?" is a crucial topic to answer in order to arrive at an usable quantitative definition of temperature. I'll discuss this quickly below, but I recommend reading K&K (linked below) for a complete, clear, and full discussion of this crucial result.
With a total energy E, S can exist in a variety of internal states (microstates). S3 atoms can share total energy in a variety of ways. Assume there are N distinct states. In the two subsystems S1 and S2, each state corresponds to a specific division of the total energy. E1 in S1 and E2 in S2 are two microstates that correspond to the same division. Only one division of the energy will occur with any substantial probability, according to a simple counting argument. It's the one containing the overwhelming majority of microstates for the entire S3 system. That figure, N(E1,E2) is just the product of the number of states allowed in each subsystem, N(E1,E2) = N1(E1)*N2(E2), and, since E1 + E2 = E, N(E1,E2) reaches a maximum when N1*N2 is stationary with respect to variations of E1 and E2 subject to the total energy constraint.
For convenience, physicists prefer to frame the question in terms of the logarithm of the number of microstates N, and call this the entropy, S. You can easily see from the above analysis that two systems are in equilibrium with one another when (dS/dE)1 = (dS/dE)2, i.e., the rate of change of entropy, S, per unit change in energy, E, must be the same for both systems. Otherwise, energy will tend to flow from one subsystem to another as S3 bounces randomly from one microstate to another, the total energy E3 being constant, as the combined system moves towards a state of maximal total entropy. We define the temperature, T, by 1/T = dS/dE, so that the equilibrium condition becomes the very simple T1 = T2.For most systems, this statistical mechanical definition of temperature corresponds to your intuitive sense of temperature. T is always positive as long as dS/dE is always positive. Adding energy to a common scenario, such as a group of free particles or particles in a harmonic oscillator potential, always increases the number of accessible microstates, and this increase accelerates as total energy grows. So, starting at 0, temperature rises with increasing energy, asymptotically approaches positive infinity as energy rises.
Step II: What is "Negative Temperature"?
The fact that entropy increases monotonically with energy is not shared by all systems. For particular ranges of energies, the number of viable microstates, or configurations, actually decreases as energy is added to the system. Consider an ideal "spin-system," which consists of N atoms with spin 1/2 on a one-dimensional wire. The atoms are unable to move freely from their wiring locations. Spin-flip is the only degree of freedom they have: a particular atom's spin can point up or down. In a strong magnetic field, the total energy of the system B, pointing down, is (N+ - N-)*uB, where u represents each atom's magnetic moment and N+ and N- represent the number of atoms with spin up and down, respectively. E is zero when half of the spins are up and half are down, according to this definition. When the majority is down, it is negative; when the majority is up, it is positive. The lowest possible energy state, all the spins pointing down, gives the system a total energy of -NuB, and temperature of absolute zero. There is only one configuration of the system at this energy, i.e., all the spins must point down. The entropy is the log of the number of microstates, so in this case is log(1) = 0. If we now add a quantum of energy, size uB, to the system, one spin is allowed to flip up. There are N possibilities, so the entropy is log(N). If we add another quantum of energy, there are a total of N(N-1)/2 allowable configurations with two spins up. The entropy is increasing quickly, and the temperature is rising as well.
However, for this system, the entropy does not go on increasing forever. There is a maximum energy, +NuB, with all spins up. There is just one microstate at this maximal energy, and the entropy is zero once more. We can enable one spin down by removing one quantum of energy from the system. There are N microstates possible at this energy. As the energy is reduced, the entropy increases. In reality, for total energy zero, i.e. half of the spins up, half down, the highest entropy occurs.
When a result, we've developed a system in which, as additional energy is added, the temperature rises to positive infinity as maximum entropy is approached, with half of all spins up. As the energy climbs toward maximum, the temperature becomes negative infinite, decreasing in magnitude until it reaches zero, but always remaining negative. It is hotter when the system has a negative temperature than when it has a positive temperature. Heat will flow from the negative-temperature system to the positive-temperature system if two copies of the system, one with positive and one with negative temperature, are placed in thermal contact.
Step III: What Does This Have to Do With the Real World?
Is this system a fascinating invention of malevolent theoretical condensed matter physicists, or can it ever be achieved in the actual world? In addition to spin, atoms always have other degrees of freedom, which makes the total energy of the system limitless upward due to the atom's translational degrees of freedom. As a result, a particle can only have negative temperature in specific degrees of freedom. It makes sense to define a collection of atoms' "spin-temperature" if one condition is met: the coupling between atomic spins and other degrees of freedom is sufficiently weak, and the coupling between atomic spins is sufficiently strong, that the timescale for energy to flow from the spins into other degrees of freedom is very large compared to the timescale for thermalization. The temperature of the spins should be discussed independently from the temperature of the atoms as a whole. Under the case of nuclear spins in a strong external magnetic field, this criterion is easily met.
Radio frequency approaches can be used to boost nuclear and electron spin systems to negative temperatures. The articles listed below, as well as the references therein, contain many experiments in negative temperature calorimetry, as well as applications of negative temperature systems as RF amplifiers, etc.
Key takeaway:
- Absolute zero, often known as zero kelvins, is a temperature of 273.15 degrees Celsius (-459.67 degrees Fahrenheit) that indicates the point on a thermometer where a system has reached its lowest potential energy, or thermal motion. However, there is a catch: absolute zero is unattainable.
- The Celsius scale, often known as centigrade, is a temperature measurement scale. The degree Celsius (°C) is the measuring unit. It is one of the world's most widely used temperature units.
- The particles stop moving and all chaos vanishes at zero kelvin (minus 273 degrees Celsius). On the Kelvin scale, nothing can be colder than absolute zero.
References:
1. Statistical Mechanics, B.K.Agarwal and Melvin Eisner (New Age Inter-national)-2013
2. Thermodynamics, Kinetic Theory and Statistical Thermodynamics: Francis W.Sears and Gerhard L. Salinger (Narosa) 1998
3. Statistical Mechanics: R.K.Pathria and Paul D. Beale (Academic Press)- 2011




