Unit - 4
Thermodynamic and Kinetic Aspects and Reaction Mechanism of Metal Complexes
Q1) Define the Stability of Metal Complexes.
A1) The ability of a metal complex to persist in favourable conditions and have a long shelf life is referred to as "stability." The term "stability of metal complex" cannot be applied universally because the complex may be stable to one reagent/condition but decompose in the presence of another reagent/condition. Metal complexes can be explained in terms of their thermodynamic and kinetic stability. A metal complex, on the other hand, is said to be stable if it does not react with water, causing the system's free energy to decrease, or thermodynamic stability. If the complex reacts with water to form a stable product and there is a known mechanism for the reaction, it is said to have kinetic stability. For example, the system might not have enough energy to break a strong bond, but once it is broken, a new bond that is stronger than the old one can be formed. The bond dissociation energy, Gibbs free energy, standard electrode potential, pH of the solution, and rate constant or activation energy for substitution reactions are used to determine the stability of a complex compound in aqueous solution.
Q2) Explain the Stability of Thermodynamics.
A2) The term "thermodynamic stability" refers to a complex's tendency to exist in equilibrium conditions. It determines how much of the complex will form or be converted into another complex at the point of equilibrium. To put it another way, thermodynamic stability of complexes is proportional to the metal-ligand bond energies and measures a metal ion's proclivity to form a specific metal complex. Complexes' thermodynamic stability is represented by the formation constant. The formation constant, also known as the stability constant, is the equilibrium constant for determining the formation metal composition.
Metal complexes are usually made in aqueous solution rather than in gaseous form from their respective starting materials. When a metal cation is hydrated in an aqueous solution, an aqua complex of the type forms. [M(H2O)x]n+ is a compound of [M(H2O) and [M(H2O) When a ligand replaces the water molecule in the aqua complex ion, a new metal complex forms and equilibrium is established.
Q3) Explain relation between thermodynamic and kinetic stabilities.
A3) The thermodynamic and kinetic descriptions of metal complexes' stability and reactivity, respectively, are used. Stable and unstable are thermodynamic terms, whereas labile and inert are kinetic terms. A metal complex is considered labile if it reacts in 1 minute at 25°C, and inert if it takes longer.
The energy change that occurs while starting materials are converted to products, referred to as G for the reaction, is referred to as thermodynamic stability.
The equation G = HTS = RTlnK gives the change in free energy, where S is entropy, H is enthalpy, and K is the reaction's equilibrium constant. The ability of a metal complex to undergo ligand substitution reactions is referred to as kinetic stability. Labile complexes are those that undergo a fast ligand substitution reaction, while inert complexes are those that take a long time. Complexes' thermodynamic and kinetic stabilities are often parallel, but they are not always. Because it undergoes ligand substitution reaction very quickly, [Ni(CN)4]2 is a good example of a thermodynamically stable and kinetically inert complex. The cobalt complex [Co(NH3)6]3+, on the other hand, is kinetically inert but thermodynamically unstable. In acidic solution, the complex [Co(NH3)6]3+ was found to decompose at a rate of 1025, indicating that it is thermodynamically unstable. When the complex is kept in acidic solution for several days, however, no ligand substitution reaction occurs, indicating that it is kinetically inert. The stability of a complex is largely determined by the conditions, as shown in the preceding two examples, and it is always advisable to mention the conditions, such as pH, temperature, and so on, when discussing the complex's stability. To summarise, a stable complex does not have to be inert, and an unstable complex does not have to be labile.
Take a look at the [Ni(CN)4]2, [Mn(CN)6]3, and [Cr(CN)6]3 complexes. Although all of the complexes are thermodynamically stable, they behave differently kinetically. When carbon-14-labeled cyanide ions react in solution with metal complexes, the rate of exchange can be measured. It proves that not all stable complexes are inert and vice versa by showing that [Ni(CN)4]2 is labile, [Mn(CN)6]3 is less labile, and [Cr(CN)6]3 is inert.
Q4) Describe Difference between thermodynamic and kinetic stability.
A4) To comprehend the distinction between kinetic and thermodynamic stability, you must first comprehend potential energy surfaces and how they relate to a system's state.
A potential energy surface depicts a system's potential energy as a function of one or more of the system's other dimensions. The other dimensions, in most cases, are spatial in nature. Chemical systems' potential energy surfaces are typically complex and difficult to depict. Fortunately, we can simplify things by beginning with simple 2-d models and then expanding our knowledge to the generalised N-d case.
So let's begin with the most basic type of potential energy: gravitational potential energy. This is simple for us because we live on the planet and are constantly influenced by it. If given the chance, things tend to move from higher to lower places, which we have developed an intuitive sense of. You can probably guess that the rock will eventually roll downhill and come to rest at the valley's bottom.
However, you intuitively understand that it will not move unless something causes it to do so. To put it another way, it requires some kinetic energy to get started.
By slightly altering the surface, I could make it even more difficult for the rock to move.
The potential energy surface's first valley is known as a local minimum. This means that the first derivative of potential energy with respect to position is zero in mathematical terms:
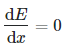
And the second derivative is positive:
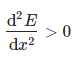
To put it another way, the slope is 0 and the shape is concave upward (or convex).
The global minimum is in the deep valley to the right (at least as far as we can tell). The mathematical properties are the same, but the energy magnitude is lower – the valley is deeper.
If you combine all of this (and are willing to accept a little anthropomorphization), you could say that the rock wishes to reach the global minimum, but its ability to do so is limited by the amount of kinetic energy it possesses.
Q5) Explain the condition of Thermodynamically favorable but kinetically unfavorable.
A5) Take a look at how much easier it is to understand. This means that the products have a lower free energy than the reactants and are therefore more stable. The reactants "wish" to be converted into the products as a result of this. Graphite and diamond, for example, are both carbon compounds, but graphite has a lower free energy than diamond. As a result, diamond desires to become graphite. When your skin is washed, it wants to dissolve in the soap. The dissolution reaction's products (your skin dissolved in soap) are more stable in this case than the reactants (your undissolved skin and the soap separately).
The examples above were chosen because, despite the fact that the reaction should work (the products are more stable than the reactants, and nature always tries to make things as stable as possible), it doesn't. When you wash your hands, the diamond on your ring finger is safe. The reaction is slow, even though it is thermodynamically favourable. It's simply too difficult to break all of the diamond's bonds and re-form them in a new, more stable graphite configuration. To understand why this is, return to the first reaction diagram, which shows the hump between reactants and products. That hump represents how difficult it is to get a reaction to happen in a reasonable amount of time. The hump for graphite conversion in diamond is quite high. The reaction does not proceed thermodynamically because it is kinetically unfavourable.
Q6) What are the properties of Thermodynamic?
A6)
• Thermodynamic properties are characteristics of a system that can be used to specify its state. Thermodynamic properties can be either extensive or intensive.
• The properties of an intensive system are those that are independent of the amount of matter in the system. Temperature and pressure are two properties that necessitate a great deal of thought.
• The system's mass determines the value of extensive properties. Volume, energy, and enthalpy are all important properties to consider.
Q7) What are the factors that affect Stability of Thermodynamics?
A7) The stability of a coordination compound depends upon the following factors:
(1) Charge density of the central metal ion:
(i)I Charge density is defined as the ratio of charge magnitude to metal ion radius.
(ii) The charge density of a complex influences its stability.
(iii) For example the ionic radii of divalent ions 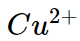 and
and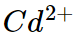 are 69 pm and 97 pm respectively hence charge density of
are 69 pm and 97 pm respectively hence charge density of 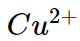 ion is higher than that of
ion is higher than that of  . Therefore the complex
. Therefore the complex  is more ions is,
is more ions is,
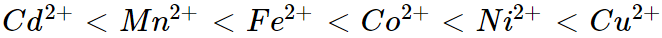
(2) Nature of ligands: The ligands are Lewis bases because they donate electron pairs to the complex's central metal ion, forming coordinate bonds. As a result, the greater the ligands' basic strength, the greater their tendency to donate electrons, and thus the complex's stability.
CN-, for instance, is more basic than NH3. As a result, cyano complexes of a metal ion are more stable than ammine complexes of the same metal ion.
Q8) Enlist and explain Nucleophilic Substitution Reactions.
A8)
(a) What are nucleophiles?
Nucleophiles are species that are strongly attached to the region of a positive charge, whether in the form of an ion or a molecule. The presence of negative ions on a molecule is said to be fully charged. Cyanide ions, water, hydroxide ions, and ammonia are common examples of nucleophiles.
(b) What is Nucleophilic substitution reaction?
In organic chemistry, a nucleophilic substitution reaction occurs when a nucleophile attaches to the other substance's positively charged atoms or molecules.
(c) Mechanism of Nucleophilic substitution reaction:
This paper discusses two mechanisms of nucleophilic substitution reactions. The SN1 reaction and the SN2 reaction, where S stands for chemical substitution, N for nucleophilic, and the number represents the reaction's kinetic order.
Q9) Describe the Mechanism of SN2 Reaction.
A9) The leaving group is eliminated while the nucleophile is added simultaneously in this reaction. The nucleophile is easily accessible to the central carbon atom in SN2.
A number of factors influence the rate of SN2 reactions. Below, we'll go over them.:
• The numerical value 2 in SN2 indicates that the rate of reaction is affected by two concentrations of substances: nucleophile and substrate.
• Rate = k [Sub][Nuc] is the rate equation for the reaction mentioned above.
• Because aprotic solvents like acetone, DMSO, or DMF do not add H+ ions to the solution, they are best for the SN2 reaction.
• The complex turns green when concentrated hydrochloric acid is added to the solution, yielding the complex [CuCl4]2+. The ligand exchange rate in inert complexes is very slow, and the ligands are difficult to exchange. The cobalt complex [Co(NH3)6]3+, for example, has a slow reaction at room temperature and no reaction at all. The aqueous solution was infused with HCL. When the aqueous solution of the complex was heated with 6M hydrochloric acid, only one NH3 ligand was found to be replaced by Cl ligand.
Q10) What is Decomposition Reactions?
A10) When a complex substance is broken down into two or more simpler substances, it is called a decomposition reaction. The general form of a decomposition reaction is ABA+B.
Most decomposition reactions require a source of energy, such as heat, light, or electricity.
Compounds with only two elements are known as binary compounds. The simplest type of decomposition reaction occurs when a binary compound decomposes into its constituent elements. Mercury (II) oxide, a red solid, decomposes into mercury and oxygen gas when heated.
2HgO(s)+2Hg(l)+O2(g)
The reaction is classified as a decomposition reaction even if one or more of the products is still a compound. Metal carbonates decompose into metal oxide and CO2 when exposed to oxygen. When calcium carbonate is exposed to air, it decomposes into calcium oxide and carbon dioxide.
CaCO3(s)→CaO(s)+CO2(g)
When heated, metal hydroxides decompose into metal oxides and water. Sodium hydroxide decomposes to produce sodium oxide and water.
2NaOH(s)→Na2O(s)+H2O(g)
Q11) Explain the Single-Replacement Reactions and Double-Replacement Reactions simultaneously.
A11) Single-Replacement Reactions:
When one element in a compound replaces another similar element in a single-replacement reaction, also known as a single-displacement or substitution reaction. In its most basic form, a single-replacement reaction is written as A+BCAC+B.
In this reaction, element AA is a metal that replaces element BB, which is also a metal. The general equation when a nonmetal replaces another nonmetal in a compound is:
Y+XZ→XY+Z
Y is a nonmetal and replaces the nonmetal Z in the compound with X.
Magnesium, unlike copper, is a more reactive metal. In an aqueous solution of copper (II) nitrate, a strip of magnesium metal replaces the copper. Aqueous magnesium nitrate and solid copper metal are the products of the reaction.
Mg(s)+Cu(NO3)2(aq)→Mg(NO3)2(aq)+Cu(s)
Many metals react readily with acids, producing hydrogen gas as one of the byproducts. Aqueous zinc chloride and hydrogen are created when zinc reacts with hydrochloric acid.
Zn(s)+2HCl(aq)→ZnCl2(aq)+H2(g)
Double-Replacement Reactions
The positive and negative ions of two ionic compounds exchange places to form two new compounds in a double-replacement reaction, also called double-displacement. A double-replacement reaction takes the following general form:
AB+CD→AD+CB
B and D are negatively charged anions, while A and C are positively charged cations in this reaction. Between substances in aqueous solution, double-replacement reactions are common. A solid precipitate, a gas, or a molecular compound such as water is usually one of the products of a reaction.
When the cations from one reactant combine with the anions from the other to form an insoluble ionic compound in a double-replacement reaction, the result is a precipitate. The following reaction occurs when aqueous potassium iodide and lead (II) nitrate solutions are mixed.
2KI(aq)+Pb(NO3)2(aq)→2KNO3(aq)+PbI2(s)
Q12) Explain the Ligand Substitution Reactions.
A12) Transition metal complexes can swap ligands, which is important in their synthesis, stereochemistry, and catalytic chemistry. Reaction kinetics are inextricably linked to the mechanisms of chemical reactions. Transition metal reactions have mechanisms that are inferred from experiments that look at the concentration dependence of the incoming and outgoing ligands on the reaction rate, the detection of intermediates, and the stereochemistry of the reactants and products, similar to organic chemistry.
Kinetics vs. Thermodynamics. It's important to remember the difference between thermodynamics and kinetics when thinking about transition metal complex reactions. Take the formation of the square planar tetracyanonickelate complex as an example:
Ni2+(aq)+4CN−(aq)=[Ni(CN)4]2−(aq)K(eq)≈1030M−4
The equilibrium of [Ni(CN)4]2- is very stable thermodynamically, implying that the equilibrium above is very far to the right. The complex, on the other hand, is labile kinetically, which means it can rapidly exchange its ligands. The exchange of a 13C labelled CN- ion with a bound CN- ligand, for example, occurs in tens of milliseconds.
[Ni(CN)4]2−(aq)+⋅CN−(aq)⟶[Ni(CN)3(⋅CN)]2−+CN−(aq)kexchange≈102M−1s−1
A compound can be thermodynamically unstable but kinetically inert, which means it takes a long time to react. The [Co(NH3)6]3+ ion, for example, is unstable in acid, but it takes about one week at room temperature to complete its hydrolysis reaction with concentrated HCl:
[Co(NH3)6]3+(aq)+6H3O+(aq)⟶[Co(H2O)6]3+(aq)+6NH+4(aq)Keq≈1030
Transition metal complexes were classified as labile if their reaction half-life was one minute or less, and inert if they took longer to react, according to Henry Taube, who studied the mechanisms of ligand exchange reactions in simple test tubes. The dynamic range of ligand substitution rates spans at least 15 orders of magnitude. The Taube definition of lability is useful for classifying reactions with low and high activation energies on the timescale of most laboratory experiments.
Q13) Define Ligand Substitution Mechanisms.
A13) For an MLn complex undergoing ligand substitution, there are essentially three different reaction mechanisms:
A MLn complex loses a ligand to form an MLn-1 intermediate in the dissociative mechanism, and the incoming ligand Y reacts with the MLn-1 fragment: Ln1ML+L, k1L, k1Ln1M+Y, k2Ln1MY
The mechanism for ligand substitution on an octahedral ML6 complex is shown below. In this example, the intermediate state includes a trigonal bipyramidal ML5 fragment as well as free L and Y ligands.
The concentration of Y has no effect on the rate of reaction if the rate-determining step is the dissociation of L from the complex, resulting in the first-order rate law.
Illustration of the dissociative ligand substitution mechanism for an ML6 complex. The reaction energy profile is shown at the right.
Rate=k1[MLn]
This reaction is first order in ML6 and zero order in Y in the case of an octahedral complex, but only if the highest energy transition state occurs before the formation of the ML5 intermediate. The rate law becomes more complicated when the two transition states have similar energy levels (as in the animation to the right). We can simplify the problem in this case by assuming that the MLn intermediate has a low steady-state concentration. The rate law that emerges as a result of this process is:
Rate=k1k2[Y][MLn]k−1[L]+k2[Y]
When k2[Y] >> k-1[L], the rate law is reduced to the simpler first-order rate law. Because dissociation of a ligand is required to form the transition state, the entropy of activation is always positive in the dissociative mechanism.
In the associative mechanism, the incoming ligand Y attacks the MLn complex, forming a transient MLnY intermediate, which then loses a ligand L, resulting in the MLn-1Y product.
Associatively substituted complexes are usually coordinatively unsaturated or contain a ligand that can change its bonding to the metal, such as a nitric oxide ligand that can bend or change its hapticity (NO). The associative pathway is preferred in homogeneous catalysis because the binding event, and thus the reaction selectivity, is dependent not only on the nature of the metal catalyst but also on the molecule involved in the catalytic cycle.
Vaska's complex (IrCl(CO)[P(C6H5)3]2) and tetrachloroplatinate are two examples of associative mechanisms found in the chemistry of d8 square planar metal complexes (II). These compounds (ML4) bind the incoming (substituting) ligand Y to form pentacoordinate intermediates ML4Y, which dissociate one of their ligands in a subsequent step. The Berry pseudorotation provides a low-energy pathway for all ligands to sample both the equatorial and axial sites, despite the fact that the incoming ligand is initially bound at an equatorial site. According to the principle of microscopic reversibility, ligand dissociation must start at an equatorial location. When Y is dissociated, no reaction occurs, but when L is dissociated, net substitution occurs, yielding the d8 complex ML3Y. Choosing a rate is usually the first step. As a result, the activation entropy is negative, indicating that the transition state has increased order. Second-order kinetics governs associative reactions: the rate at which product appears is determined by the concentrations of both ML4 and Y.
Q14) Define the Trans Effect.
A14) The Trans Effect, which is connected with the associative mechanism, controls the stereochemistry of certain ligand substitution reactions.
The labilization (increased reactivity) of ligands that are trans to other ligands, also known as trans-directing ligands, is referred to as the trans effect. Trans ligand labilization, which is most noticeable in square planar complexes but also seen in octahedral complexes, is thought to be caused by electronic effects. The cis effect is most commonly observed in octahedral complexes.
In addition to the kinetic trans effect, trans ligands have an impact on the molecule's ground state, the most notable of which are bond lengths and stability. Some authors prefer the term trans influence to distinguish this from the kinetic effect, while others prefer more specific terms like structural trans effect or thermodynamic trans effect to differentiate this from the kinetic effect.
The discovery of the trans effect is credited to Ilya Ilich Chernyaev, who recognised and named it in 1926.
The intensity of the trans effect (as determined by the rate of substitution of the trans ligand) is determined by the following sequence:
Weak field ligands are not trans-directing, but strong field ligands are.
The synthesis of cisplatin and its trans isomer is a well-known example of the trans effect. The first NH3 ligand is randomly added to one of four equivalent positions starting with PtCl42. Due to the fact that Cl has a greater trans effect than NH3, the second NH3 is added trans to a Cl and thus cis to the first NH3..

If, on the other hand, one starts from Pt(NH3)42+, the trans product is obtained instead:
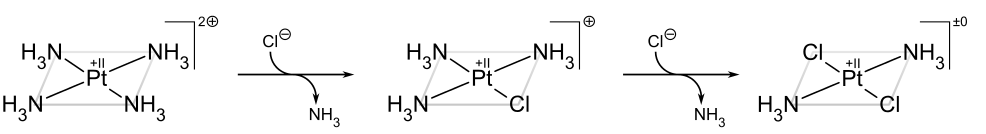
The trans effect in square complexes can be explained using the associative mechanism described above, which passes through a trigonal bipyramidal intermediate. In general, ligands with high acidity (as in phosphines) or low-ligand lone-pair–d repulsions (as in hydride) prefer the intermediate's more -basic equatorial sites, which prefer the intermediate's more -basic equatorial sites. The second equatorial position is taken by the incoming ligand. Because the departing trans ligand occupies the third and final equatorial site, the kinetically preferred product is the one in which the ligand trans to the one with the greatest trans effect is removed.
• The interchange mechanism is similar to the associative and dissociative pathways except that no distinct MLnY or MLn-1 intermediate is formed. Nucleophilic substitution via the SN2 pathway at a tetrahedral carbon atom is analogous to this coordinated mechanism in organic chemistry. The interchange mechanism is classified as either associative (Ia) or dissociative (Id) depending on how important M-Y and M-L bonding are in the transition state (Id). If a strong M-Y bond forms during the transition state, the mechanism is Ia. The mechanism is Id if the weakening of the M-L bond is more important in achieving the transition state.
• The exchange of bulk and coordinated water in [V(H2O)6]2+ is an example of the Ia mechanism. The slightly more compact ion [Ni(H2O)6]2+, on the other hand, uses the Id mechanism to exchange water.
Q15) Define the Polarization theory.
A15) The polarisation (effect of -bonding) of a ligand is proportional to its Trans effect, according to this theory. There is no trans effect because the overall dipole generated by the [Pt(X)4] type complex is zero. In the [PtL(X)3] type complex, the dipole across the opposite X cancels each other, so there is no trans effect. Because the dipoles of the two ligands X and L are opposite, they are not cancelled (trans). Due to its larger size, L has a higher polarisability than X. Pt (II) deforms due to L's induced dipole, resulting in more effective bonding and higher bond energy in L-Pt compared to Pt-X trans to it. As a result, the Pt-X bond has become weaker, while the Pt-L bond has become longer. The rate of X reater polarisability, L has a greater trans effect than X.
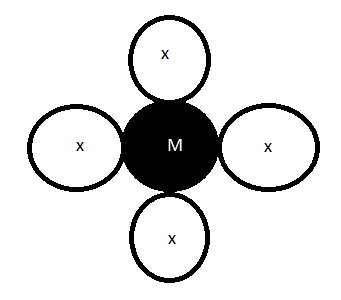
Figure: Electrostatic polarization in Pt(II) complex
M-X bonds become weaker and longer as the Trans effect of L increases. With increasing polarisability, the Trans effect of ligands that are unable to form metal ligand-bonds increases, e.g.
Cl-< Br-< I-
Polarizability increases
Trans effect increases
B. The π- bonding Theory
Metal-ligand -bonds ( d-d or d-p bond) can be formed when ligands with vacant or* orbitals accept electrons from filled metal orbitals. CO, CN-, C2H4, PR3, and other ligands are examples of -bonding ligands. The ability of -bonding ligands to form metal–ligand–bonds increases as their Trans directing ability increases. The formation of the -bond, for example, increases the electron density on the Pt-L bond while decreasing Trans of L in the [PtLX3] type complex. As a result of these findings, the Pt-X bond is weaker than the Pt-L bond, and thus substitution occurs from X to L.

The -acceptor ligands increase the reaction intermediate's stability (i.e. trigonal bi pyramidal). Because of the poor bonding of this bond with Pt, the incoming Y enters in the direction of the Pt-X bond, trans to L. (II). The d-p and d-d bond substitution increases as the Pt-X bond weakens and becomes Pt-L. Due to its g formations.
Q16) State the Coordination Rates and the Activated State.
A16) For various concentrations and initial temperatures, the steady-state and transient behaviours are shown. At different wavelengths, distinct transient behaviour is observed. For a probing wavelength of 480 nm, a decay is observed, indicating the depletion of the initial population of hexa aqua complexes at concentration-dependent rates. Depending on the ligand concentration, a rise or decay is observed for detection wavelengths between 550 and 630 nm. The accumulation or depletion of intermediate populations determines the behaviour in this region. The formation of the final chloro cobalt complexes causes a rise in absorption at probing wavelengths of 630 nm.

Octahedron and tetrahedron structures and the spectra of the cobalt complexes. Spectroscopic evidence of equilibrium shift at different sample temperatures (Middle) and ligand concentration (Bottom) are shown with the two structures (Top).

At various wavelengths, representative transients for cobalt complexes. (A) Decay curves, which show the rate of depletion of the initial reactants, the hexa aqua octahedral species, as a function of ligand concentration. (B) The formation or depletion of intermediate states as represented by rise or decay curves. (C) The final product, the tetra chloro tetrahedral species, is formed by rising curves. (D) The observed rates' amplitude has distinct peaks and a blue shift from kinital to kfinal behaviour.
To improve quantification, we used a multiphase exponential model to fit the datasets of all transients recorded under various conditions, and we were able to obtain consistent amplitudes and rate constants (shared as global parameters). The findings show that three distinct components exist: kinitial1 (200–1,000 ps), klimit1 (1–10 ns), and kfinal1 (200–1,000 ps); the components differ, as discussed below. Only samples with a low to medium [Cl]/[Co2+] ratio (5.0) show the first-rate process, kinitial, which represents the relatively fast octahedron configuration substitution process. Following this initial process, a slower rate step with an amplitude that peaks between the kinitial and kfinal processes indicates that the octahedral–tetrahedral structural change is the sequential substitution's rate-limiting step, as discussed below. The study of this amplitude's concentration dependence reveals that the amplitude shifts significantly with concentration. Finally, at high [Cl]/[Co2+] ratios (>12), a relatively fast process with rate constant kfinal is recovered, which is assigned to the tetrahedral chlorocobaltic product substitution rate. (Eq. 2d):
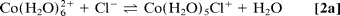
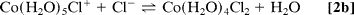
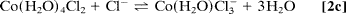
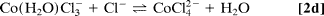
The concentration dependence of the reaction rates is plotted in which follows a linear relationship with the chloride activity (aCl);k = kligand aCl + kwater. The rate constants of ligand (chloride) and water attack, respectively, are represented by the slope (kligand) and intercept (kwater). The average chloride activity, aCl, is given by aCl = [Cl]100.19[Cl]0.53 (10) and is significant for high concentration solutions (>5 M). The substitution in octahedral complexes was expected to follow a dissociative interchange pathway (15–17), in which the rate of leaving water molecule(s) becomes a deciding step. The observed linearity over a wide range and up to 13 M, on the other hand, suggests that the chloride anation is the bimolecular process' rate-limiting step. As a result, it's a mechanism of interchange in which the bond-forming association is crucial.
Q17) Define Ionic Diffusion and Collisions. State its Mechanism and Conclusion.
A17) Because the reactions involve bimolecular collisions in solution, the effect of diffusion should be considered. Debye's theory can be used to estimate the diffusion of two ions with charges Z1 and Z2 at low electrolyte concentrations.,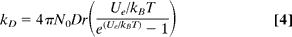
Where D = D1 + D2, D1 and D2 are the ions' diffusion coefficients, Ue = z1z2e2/4r is the electrostatic potential, N0 is Avogadro's number, and r is the distance between the two ions' closest approach (19). The diffusion coefficient for Cl in water is estimated to be 4.4 105 cm2s1 using the Stokes–Einstein equation [Do = kBT/6r]; the contribution of the complex to the diffusion rate can be ignored due to its size being much larger than the chloride ion. . In the range of 1–50 109 M1s1, the typical diffusion-controlled rate is estimated. It is faster by one to two orders of magnitude than observed reaction rates. As a result, the process must account for collision efficiency as well as the geometrical constraint that exists between the large complex and the small chloride ion (20).
Mechanism
The following picture of a two-step process can be drawn from the above results. By electrostatic interactions, the ligand diffuses into the second coordination sphere and forms an ion pair. This is a pre association process based on the Eigen and Wilkins concept (21, 22). When successful collisions occur, the reaction occurs. Because the cobalt complexes become more negatively charged and less sterically crowded as the reaction progresses toward the product, we would expect the water attack kinetics to accelerate while the chloride attack kinetics to slow. Indeed, the rate constants' observed trend supports this simple picture. The rate-limiting step is structural conversion from octahedral to tetrahedral coordinate, where chloride is anated to the complex in a stepwise manner along the reaction coordinate. The large desolvation enthalpy (two extra water molecules are removed when the substitution is coupled with the structural change) as well as possible entropic penalty contributions from rearrangement of the activated complex configuration could all contribute to the dynamical bottleneck. Water exchange is orders of magnitude slower (or longer) without the help of ligands.
Conclusion
The ultrafast T-jump study shows that the substitution of the Co(II) ion in aqueous solution occurs in multiple steps, with the octahedral-to-tetrahedral structural change being the rate-limiting step. The reaction has the characteristics of an associative interchange mechanism, in which the ion in the coordination shell is essential for water liberation and structural changes. In theoretical simulations using transition path sampling, the “simultaneous” interaction of the entering and departing groups with the metal centre has been elucidated for similar octahedral complexes of Cr (23). The activation energy of entering and departing ions is significantly reduced due to favourable interactions with the metal centre. Only one rate, kinitial, was observed along the reaction coordinate of the Co system, despite the fact that there are two initial steps along the reaction coordinate. This suggests that the kinitial process may be multifaceted. The “abnormal” negative activation enthalpy deduced from kinitial vs. T supports the hypothesis that this multistep process includes a fast preequilibrium step.
The study of the dynamics of metal complexes associated with biomolecules, such as those involving proteins or DNA, is a natural extension of our research using this ultrafast T-jump method.
Q18) Explain the Substitution processes with DNA constituents and amino acid in Cobalt Complex Preparation.
A18) In biological systems, Zn(II) and Cu(II) prefer O-carboxylate, carbonyl, and N-imidazole donor bioligand due to their square pyramidal structure. N1 and N7 allow inosine-5'-monophosphate (5'-IMP) and guanosine-5'-monophosphate (5'-GMP) to coordinate with metal ions. The N7 atom is deprotonated in our experiment (pH 7.38). The nucleotide's 5'-monophosphate residue (pK a 6) is also expected to be partially deprotonated at this pH. 1H and 31P NMR spectroscopy were used to confirm binding to Zn(II) through the N7 position of 5'-GMP in a neutral or weakly acidic medium. During the investigation of transition metal ions' hydrolytic cleavage ability, the possibility of Cu(II) complex interaction with oxygen from phosphate residue in DNA was confirmed, owing to steric hindrance..
According to a comparison of the reactivity of the two complexes, [CuCl2(terpy)] is at least 2-10 times more reactive than [ZnCl2(terpy). Both complexes with 5'-IMP and 5'-GMP have second-order rate constants for the first reaction step that are of the same order of magnitude. The similar reactivity of 5'-IMP towards [CuCl2(terpy)] for both reaction steps could be due to a decrease in the electronic density on the copper(II) centre caused by the tridentate chelate, 2,2′:6′,2”-terpyridine's -acceptor ability, resulting in both chloride being equal for parallel substitution routes. In comparison to cisplatin-based drugs, coordination's versatility may allow for the treatment of various cancer types with varying toxicity. According to the literature, there are few studies of ligand-substitution reactions between Zn(II) and Cu(II) complexes and biologically relevant nucleophiles, so bioligand reactivity was compared to kinetic data for substitution reactions of the [PtCl(terpy)]+ complex [17b). By comparing the order of reactivity of biomolecules in substitution reactions with Pt(II), it was discovered that the order of reactivity of biomolecules in substitution reactions with Pt(II) is the same, and platinum(II) complex react slightly faster with DNA constituent.
According to the hard-soft acid nature of metals, five-coordinate metal centres Zn(II) and Cu(II) prefer O-carboxylate bioligands. L-Met and DL-Asp are coordinated by O-carboxylate donor atoms, but the formation of chelate O-N-ammine has not been observed. It is well known that Cu/Zn-superoxide dismutase contains zinc(II) ion coordination to a L-aspartic acid via oxygen (O-Asp81) (SOD1). For the first step between complexes and DL-Asp, similar reactivity orders were obtained (for [ZnCl2(terpy)] k 1 = 79 ( 2 mol1 L s-1; for [CuCl2(terpy)] k 1 = 96 ( 3 mol1 L s-1). The rate constants show that the substitution of the first chloride in the Zn(II) complex by DL-Asp slows down the second substitution step by almost ten times. The substitution reactions between Zn(II) and Cu(II) complexes with L-methionine are the slowest, according to the data. At pH 7.38, reactions between the zwitterionic form of L-methionine and complexes occur (pK a1 = 2.65 and pK a2 = 9.08 are the dissociation constants for L-Met).
The similar kinetic behaviour of [ZnCl2(terpy)] and [CuCl2(terpy)] complexes suggests that the strong -acceptor ability of the tridentate chelate 2,2′:6′,2′′-terpyridine, as well as steric hindrance and electronic properties of the first coordinated nucleophiles, control the rates of nucleophilic substitution reactions. The extreme liability of [Zn(H2O)6]2+ and [Cu(H2O)6]2+ (k 298 > 107 s-1) with diffusion controlled rate constants is well known. Water exchange rates in complexes are reduced when bi-, tri-, and tetra-dentate ligands are present in the inner coordination sphere. The metal ion is forced into a different geometry by the strong ligand field. Furthermore, because of this, the rate constants of ligand-substitution reactions are reduced by a factor of 103. Based on the information in the literature, it can be concluded that the square pyramidal coordination geometry around Cu(II) has a greater impact on the rates of ligand-substitution reactions in the [CuCl2(terpy)] complex than the electronic properties and steric hindrance induced by the first coordinated biomolecules. This could explain why the rate constants for both reaction steps with 5'-IMP and DL-Asp were of similar order of magnitude/
For reactions involving L-methionine at pH 7.38, the activation parameters H and S were calculated using an Eyring equation. For both reaction steps, every available activation parameter supports an associative mechanism A or Ia. The significantly negative activation entropies indicate that bond formation appears to dominate the activation process in the studied systems. As a result, six-coordinate transition states are expected. The substitution reaction processes have an associative mechanism (A) that involves the formation of an intermediate with a higher coordination number, followed by the displacement of a weakly bound ligand. In many associative interchange reactions, there is no well-defined intermediate and bond formation between the central metal ion and the entering nucleophile takes precedence
Q19) Define Substitution reactions of aquo ions.
A19) Most metals have a hydration number of 6 (exceptions include Be2+ and Ca2+, which have a hydration number of 4; lanthanides have a higher hydration number). The substitution of bound H2O by solvent water, as shown in the equation above, is the most basic ligand substitution process. Various techniques, such as relaxation methods and NMR, have been used to investigate the kinetics of this reaction. For the vast majority of metal aquo ions, kinetic data is available, and the measured rate range is enormous, spanning 18 orders of magnitude (figure). Most metals have fast substitution, but the slow ones can be more useful in kinetic studies.
Solvent interferes with nucleophilic substitution reactions by replacing the leaving group and forming a different complex before the nucleophile attacks, which not only affects the mechanism of the reaction but also provides incorrect information about the kinetics of the reaction. The substitution reaction of the octahedral complex has been studied kinetically in an aqueous medium. The presence of acid or base in the medium can affect the rate of substitution reactions in aqueous solution.
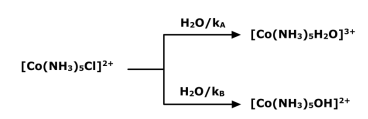
The nature of the product in hydrolysis reactions is determined by the pH of the solution; in acidic solution, the water-containing complex forms, while in basic solution, the hydroxo(HO-) complex forms. If you use the same formula for both SN1 and SN2 type reactions, the rate will be Rate of reaction = 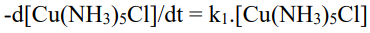 in case of SN1 reaction and
in case of SN1 reaction and 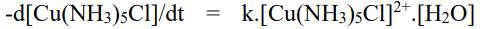 if the SN1 reaction occurs Because the concentration of water is sufficient, there will be virtually no change in water concentration. As a result, kinetic studies of acid or base hydrolysis will always lead to erroneous conclusions about the mechanism of the reaction. The hydrolysis rate constant
if the SN1 reaction occurs Because the concentration of water is sufficient, there will be virtually no change in water concentration. As a result, kinetic studies of acid or base hydrolysis will always lead to erroneous conclusions about the mechanism of the reaction. The hydrolysis rate constant 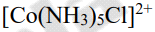 is a million times faster than an acidic solution, i.e. kB is approximately 104 to 108 times higher than kA. The next module will cover acid and base hydrolysis
is a million times faster than an acidic solution, i.e. kB is approximately 104 to 108 times higher than kA. The next module will cover acid and base hydrolysis
Q20) Eigen-Wilkins mechanism.
A20) Eigen shared the Nobel Prize in Chemistry with Norrish and Porter in 1967 for this and other work on reaction mechanisms. Interchange reactions start with a pre-equilibrium state, which leads to the formation of an encounter complex, which then rearranges to produce the products. All molecular reactions in solution that occur at rates less than the diffusion limit can be explained using the Eigen-Wilkins mechanism.
Unfortunately, under certain conditions, such as large concentrations of X, constant concentration of H2O (a very likely case), and small or large k-w, kx, etc., the rate expressions predicted by the Eigen-Wilkins mechanism can all reduce to the same expression.
As a result, the rate law is nearly useless for determining the nature of the substitution mechanism in octahedral complexes. The Id mechanism appears to be active in most octahedral complexes, according to the evidence. The evidence that supports this conclusion will be presented and discussed in the remainder of this section.
Q21) Substitution reactions of octahedral complexes.
A21) The most extensively mechanistically studied organic reaction is the substitution of ligands in octahedral metal complexes. It is crucial, and a number of significant observations and results are discovered. Substitution in octahedral systems was first investigated in aqueous solutions for classical coordination complexes. Recently, some important discoveries have been made about the mechanism by which organometallic complexes react to ligand substitution. Following a discussion of the aqueous reaction mechanisms, we will look at these organometallic complexes.
The following three sections address the following topics:
Points that are unique to Co(III), owing to the fact that it is the metal/oxidation state that has received the most research. In general, octahedral complexes undergo substitution reactions. At the octahedral level, a little organometallic substitution chemistry.




